
Figura 1: Paciente a los 4 años de edad cuando se le realizó el diagnóstico, presenta fenotipo típico de MPS I
PRESENTACIÓN DE CASOS CLÍNICOS
Mucopolisacaridosis de tipo I Hurler: Informe de un caso
Mucopolysaccharidosis I, Hurler syndrome: A case report
Milagros Amorína, Andrea Carlina y Dra. Ana Prötzelb
a. Estudiantes, Escuela de Medicina Humana, Universidad
Peruana de Ciencias Aplicadas.
b. Servicio de Genética, Hospital Nacional Edgardo
Rebagliati Martins.
Lima, Perú.
Correspondencia: Milagros Amorín: milagros_amorin@hotmail.com.
Conflicto de intereses: Ninguno que declarar.
Recibido: 1-3-2012
Aceptado: 2-5-2012
RESUMEN
La mucopolisacaridosis de tipo I (MPS I), es una enfermedad genética autosómica recesiva de origen lisosomal, caracterizada por la deficiencia de la enzima a-L-iduronidasa. La deficiencia en el catabolismo de los glucosaminoglucanos resulta en su acumulación en diferentes tejidos y órganos. La incidencia global de la MPS I es de 0,99-1,99/100 000 nacidos vivos. Existen tres presentaciones clínicas: Hurler (grave), Hurler-Scheie (moderada) y Scheie (leve). Presentamos el caso de un niño de 10 años de edad a quien se le diagnosticó MPS I, de variedad grave en el año 2006, mediante medición de la actividad enzimática de a-L-iduronidasa en leucocitos. Este caso es el único con diagnóstico confirmado y tratamiento enzimático hasta el momento, en el Perú. Presenta infecciones respiratorias recurrentes, hernia umbilical, opacidad corneal, rasgos toscos, macroglosia, hipoacusia, rigidez articular, estenosis de la válvula pulmonar leve-moderada, manos en garra, retardo mental y retraso del crecimiento. Recibe terapia de reemplazo enzimático desde el año 2008, mostrando mejoría de los síntomas viscerales, más no del daño neurológico.
Palabras clave: Mucopolisacaridosis; Síndrome de Hurler; MPS I; Trastorno genético; Terapia de reemplazo enzimático; TRE.
SUMMARY
Mucopolysaccharidosis I (MPS I) is a rare, recessively inherited, lysosomal storage disorder caused by deficiency on the enzyme a-L-iduronidase. This defect results in accumulation of heparan and dermatan sulfate in different tissues and organs due to a deficiency in the catabolism of glycosaminoglycans. The overall incidence of MPS I is 0.99-1.99/100.000 live births. There are three clinical presentations: Hurler (severe), Hurler Scheie (mild) and Scheie (mild). We report the case of a 10-years-old male patient diagnosed with Hurler syndrome, the severe presentation, 5 years ago by enzyme a-L-iduronidase activity measurement in leukocytes; with a history of recurrent respiratory infections, umbilical hernia, corneal opacity, coarse facial features, macroglossia, hearing loss, stiffness of joints, cardiac compromise, claw hands, mental retardation and stunted growth. After enzyme replacement therapy the patient has shown improvement of visceral symptoms, but the neurological damage continuous in progress.
Key words: Mucopolysaccharidosis; Hurler syndrome; MPS 1; Hurler; Genetic disorder; Enzymatic replacement therapy; ERT.
INTRODUCCIÓN
La mucopolisacaridosis de tipo I (MPS I), un
trastorno lisosomal de origen genético de tipo autosómico recesivo se debe a la deficiencia de la enzima a-L-iduronidasa. Fue descrita entre los años
1900 y 1913 por Thompson y comunicada, por primera vez, en 1917 por Hunter.1
Esta deficiencia enzimática impide el catabolismo de los glucosaminoglucanos (GAGS), lo cual
resulta en disfunción multiorgánica por acumulación progresiva de dermatán y heparán sulfato
en diferentes órganos y tejidos.2,3
Se definen tres presentaciones clínicas: Hurler (grave), Hurler-Scheie (moderada) y Scheie
(leve).4 La MPS I es una enfermedad muy poco frecuente, su incidencia global es de 0,99-1,99/100 000 nacidos vivos2; un 20% de los casos
de MPS presentan la forma atenuada.5
El síndrome de Hurler, cuya edad de aparición
de síntomas varía según la gravedad del cuadro,
se caracteriza por infecciones recurrentes del tracto respiratorio y del oído, hernia umbilical, hernia inguinal, rasgos toscos, labios gruesos, cejas
pobladas, macrocefalia, macroglosia, opacidad
corneal, retraso del desarrollo, retraso mental, hipoacusia, deformidades esqueléticas, rigidez articular y defectos cardíacos, entre otros.2,5
El objetivo de este informe es describir el cuadro clínico, la evolución y el tratamiento de la
MPS I Hurler en un paciente de 10 años de edad
del Hospital Nacional Edgardo Rebagliati Martins en Lima-Perú, debido a que es el único caso
de MPS I con diagnóstico confirmado que recibe
terapia de reemplazo enzimático en este país hasta la fecha.
HISTORIA CLÍNICA
Paciente de sexo masculino de 10 años y 5 meses de edad, producto de segunda gestación, nacido a término de parto vaginal, sin complicaciones.
Peso al nacer: 3950 g.
El primer signo de MPS I que presentó fue cifosis dorsolumbar y sufría de infecciones respiratorias recurrentes con marcada congestión nasal
que interferían con el sueño, por lo que acude al
Instituto Nacional de Salud del Niño a los 2 años
de edad. Al siguiente año surgen los demás signos: macrocefalia, facies "tosca", base nasal plana, retardo psicomotor, inadecuado desarrollo del
lenguaje, opacidad corneal, hirsutismo, rigidez articular especialmente en manos, hepato-esplenomegalia y talla corta. Se envió muestra de orina al
Japón, donde informaron GAGS urinarios elevados y definieron el diagnóstico de mucopolisacaridosis no especificada. A los 3 años, se le realiza
una adenoidectomía. A los 4 años, se le detecta un "soplo cardíaco", por lo que acude a Cardiología Pediátrica del Hospital Rebagliati y es derivado al Servicio de Genética, donde se realiza el
estudio en papel de filtro y luego la determinación enzimática de a-L-iduronidasa en leucocitos
(0,33%) y la determinación del valor enzimático en leucocitos (0,033 nmol/mg), con lo cual se
confirma el diagnóstico de MPS I a los 4 años y 7
meses de edad.
El fenotipo característico de esta patología ya
era evidente antes de empezar la terapia de remplazo (Figura 1); se aprecian facciones toscas, macrocefalia, nariz ancha con puente nasal plano
(Figura 2), hirsutismo, cifosis dorsolumbar (Figura
3), deformación de manos con apariencia de "mano en garra" (Figura 4), opacidad corneal difusa
y macroglosia.
Figura 1: Paciente a los 4 años de edad cuando se le realizó el diagnóstico, presenta fenotipo típico de MPS I

Figura 2: Puente nasal bajo
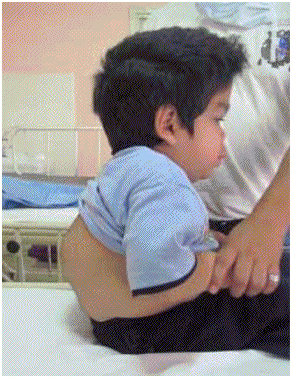
Figura 3: Cifosis dorsolumbar
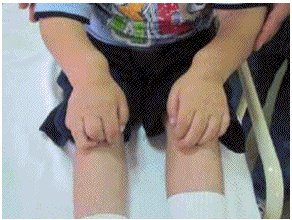
Figura 4: Manos en garra
A los 5 años y 8 meses inicia terapia de reemplazo enzimático con laronidasa (Aldurazyme®);
hubo necesidad de colocar catéter venoso central,
por ser muy difícil el acceso venoso periférico. A
las 8 semanas de tratamiento presentó marcada
mejoría de los síntomas respiratorios y el sueño
se hizo ininterrumpido. Dejó de acudir al hospital
por síndromes obstructivos bronquiales. Nunca usó métodos invasivos de ventilación durante el
sueño y la disminución de la hepatoesplenomegalia fue notable. Inicialmente, mejoró la amplitud
de movimientos de las manos, pero no cumplió una adecuada terapia física por no poder acudir
a ellas regularmente. Por otro lado, fue operado
a los 6 años de una hernia umbilical.
A un año de la TRE, se le realizó una prueba
cuantitativa de GAGS urinarios, que resulto negativa; creatinina: 37,5 mg/dl, unidades CPC/g de
creatinina: 168,4 y albician blue: 8,21 mg de GAG/mmol de creatinina. Asimismo, la única prueba
de control al año de la TRE que se realizó fue una
RNM cerebral que mostró cráneo dolicocefálico,
atrofia cerebral cortico-subcortical, sinusitis maxilar, etmoiditis y mastoiditis bilaterales.
Tras casi 4 años de tratamiento, su capacidad
visual y auditiva están muy deterioradas, la rigidez articular no ha mostrado mejoría significativa, camina con apoyo y el deterioro neurológico
continúa avanzando; sin embargo, la terapia enzimática mejoró su calidad de vida, pues el número
de hospitalizaciones por complicaciones de la enfermedad se redujo. (Figura 5)

Figura 5: Paciente a los 10 años de edad en terapia de
reemplazo enzimático
DISCUSIÓN
La MPS I tendrá mejor evolución y pronóstico
si se realiza un diagnóstico precoz e intervención
temprana. Los signos y síntomas del síndrome de
Hurler pueden ser detectados en los primeros meses de vida si se tiene en mente esta entidad. En
las formas atenuadas (Hurler-Scheie y Scheie), los
signos de sospecha pueden aparecer después del
primer año de vida.6
Existen dos opciones de tratamiento para la
MPS I: el tratamiento enzimático sustitutivo, propuesto en 1964 por De Duve; y el trasplante de
médula ósea (TMO) de donante emparentado,
propuesto en 1981 por Hobbs y cols., quienes observaron una mejoría clínica en un lactante con
síndrome de Hurler luego del TMO. Ambas opciones son hasta la actualidad los pilares del tratamiento en las enfermedades lisosomales.7
El TMO es el tratamiento de primera línea
porque, además de disminuir los síntomas viscerales, detiene el progreso del compromiso neurológico.7 Sin embargo, éste solo está indicado
cuando aún no existe deterioro cognitivo, ni daño
en el sistema nervioso central (SNC).1 Asimismo,
puede ser realizado con un donante relacionado
consanguíneo histocompatible, de preferencia un
hermano sano, o con un donante no relacionado,
preferentemente de sangre de cordón umbilical
con compatibilidad total.8,9 Cabe destacar que este trasplante debe ser efectuado antes de los dos
años de edad para evitar daños neurológicos graves e irreversibles.8 En el presente caso, el paciente
fue diagnosticado a la edad de 4 años y 8 meses,
cuando ya había compromiso importante del neurodesarrollo, por lo que no fue posible considerar
la opción del TMO.
La TRE altera la tasa de progresión de la enfermedad, es decir, puede modificar su curso natural, pero no revoca todos los síntomas.6,10 Los
síntomas cardíacos y la opacidad corneal una vez
establecidos son difíciles de tratar, pero se pueden disminuir los síntomas viscerales y mejorar
significativamente la calidad de vida y sobrevida
de los pacientes. Sin embargo, la enzima recombinante no atraviesa la barrera hematoencefálica,
por lo que el deterioro neurológico continúa progresando.1, 7,11
Finalmente, cabe resaltar que, ante un antecedente familiar de esta enfermedad, la sospecha es
sencilla y se puede confirmar el diagnóstico mediante estudios moleculares de diagnóstico prenatal o para la detección de portadores.12 Por lo
tanto, el asesoramiento genético familiar es esencial para lograr el diagnóstico precoz y realizar
medidas terapéuticas tempranas. Las expectativas terapéuticas en niños con diagnóstico temprano son buenas, ya que el tratamiento iniciado
antes de la acumulación progresiva de glucosa-minoglucanos evitará la progresión de la enfermedad y, por tanto, el compromiso neurológico.2 Pero cuando no hay antecedentes familiares, el
pediatra debe conocer la enfermedad para poder sospecharla en las consultas iniciales o en los
controles de crecimiento y desarrollo ante la presencia de infecciones óticas y respiratorias a repetición, y hernias.
En nuestro país, estamos empeñados en difundir la presencia de enfermedades poco comunes
o de baja prevalencia como ésta, que tienen un
punto crucial para su detección precoz e intervención temprana. Las enzimas de reemplazo son
medicamentos de alto costo y que no son cubiertas por las aseguradoras, por lo que el acceso es
limitado; además, existen numerosas trabas administrativas y de políticas de salud que agravan la situación de estos pacientes. Por el momento, sólo se cuenta con enzimas de reemplazo. Deberán
desarrollarse nuevas alternativas de tratamiento,
especialmente eficaces para el SNC, lo que cambiará definitivamente las expectativas de vida de
estos pacientes.
1. Mabe P. Las mucopolisacaridosis. Rev Chil Nutr 2004;31 (1):8-16.
2. Bay L. Consenso de diagnóstico y tratamiento de la mucopolisacaridosis de tipo I. Arch Argent Pediatr 2008;106(4):361-8.
3. Muenzer J, Wraith J, Clarke L. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123;19.
4. Alentado N, Cabo T, Dalmau J. Mucopolisacaridosis tipo I: evolución clínica de 2 pacientes tras 30 meses de tratamiento enzimático sustitutivo. Acta Pediatr Esp 2007;65(5):241-5.
5. Tatapudi R, Gunashekhar M, Suryanarayana Raju P. Mucopolysaccharidosis type I Hurler-Scheie syndrome: A rare case report. Contemp Clin Dent 2011;2(1):66-8.
6. Gabrielli O, Clarke L, Bruni S, Coppa G. Enzyme-replacement therapy in a 5-month-old boy with attenuated presymptomatic MPS I: 5-year follow-up. Pediatrics 2010;125(1):e183-e7.
7. Sardón O, García Pardos C, Mintegui J, Pérez Ruiz E, et al. Evolución de dos pacientes con síndrome de Hurler en tratamiento con enzima recombinante humana alfa-Liduronidasa. An Pediatr (Barc) 2005;63(1):61-7.
8. Muenzer J, Fisher A. Advances in the treatment of mucopolysaccharidosis Type I. N Engl J Med 2004;350:1932-4.
9. Staba SL, Escolar ML, Poe M, Kim Y, et al. Cord-blood transplants from unrelated donors in patients with Hurler's syndrome. N Engl J Med 2004;350:1960-9.
10. Wraith J, Clarke A, Beck M, Kolodny EM, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human a-L-iduronidase (laronidase). J Pediatr 2004;144(5):581-8.
11. Kakkis E, Muenzer J, Tiller G, Waber L, Belmont J, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001;344:182-8.
12. Rojas I, Enríquez G. Presentación de un caso clínico de mucopolisacaridosis tipo Hurler y revisión de la literatura. Rev Mex Neuroc 2001;2(3):149-60.