











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkLos tumores malignos de la pared torácica son poco frecuentes: solo representan el 7% de los tumores óseos y mesenquimales1,2. El sarcoma de Ewing (SE) es una neoplasia rara y altamente agresiva que se presenta con preferencia en adolescentes varones. Se estima que afecta anualmente a 3 personas por cada millón habitan tes3. La mayoría de los pacientes tiene la translocación t(11;22), que involucra los genes EWS y FLI1 y resulta en la formación de una proteína de fusión oncogénica3. El pronóstico del SE es pobre; sin embargo, la supervivencia ha mejorado con el advenimiento de nuevas terapias sisté micas y la incorporación de nuevas técnicas quirúrgicas y reconstructivas1,2,4. La neoadyuvancia intensiva reduce el volumen tumoral, disminuye la necesidad de radioterapia y aumenta la tasa de resección R0 (resección macro y microscópica completa)5. En este contexto, la respuesta completa al tratamiento se ha descrito de forma excep cional6. Presentamos el caso de un varón adulto con SE óseo de pared torácica en quien se obtuvo respuesta patológica completa a la terapia multimodal.
Caso clínico
Varón de 41 años, natural de Amazonas-Perú, acudió a nuestra institución por la aparición de tumor en región dorsal derecha de crecimiento rápido y 2 años de evolución. Al examen físico, en la región toracodorsal derecha se evidenció un tumor de 22 cm de diámetro, fijo a planos profundos y de consistencia dura. La resonancia magnética nuclear de pared torácica mostró una extensa lesión neoformativa dependiente de pared torácica, de bordes definidos y sin infiltración del parénquima pulmonar (Fig. 1). La histopatología de la biopsia del tumor mostró neoplasia maligna de células redondas com patible con SE. Las pruebas inmunohistoquímicas arrojaron lo siguientes resultados: panqueratina (-) , proteína S100 (-), An tígeno común leucocitario ACL (-), CD99 (+), Friend leukaemia integration-1 FLI-1 (-), sinaptofisina (-) y desmina (-). Mediante RT-PCR se detectó presencia de genes de fusión EWS FLI tipo 1, la que confirmó el diagnóstico de SE (Fig. 2 A y B).
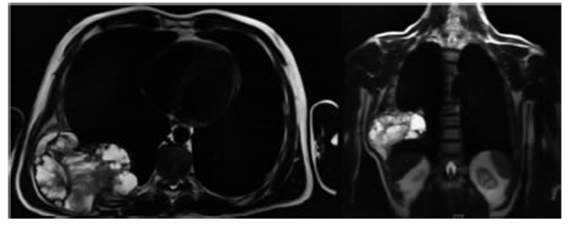
Fig. 1 Resonancia magnética nuclear de pared torácica axial y coronal: Extensa lesión neoformativa de la pared torácica de bordes definidos de 14 cm de diámetro, con señal heterogénea en secuencia ponderada en T2 con restricción patológica de la difusión
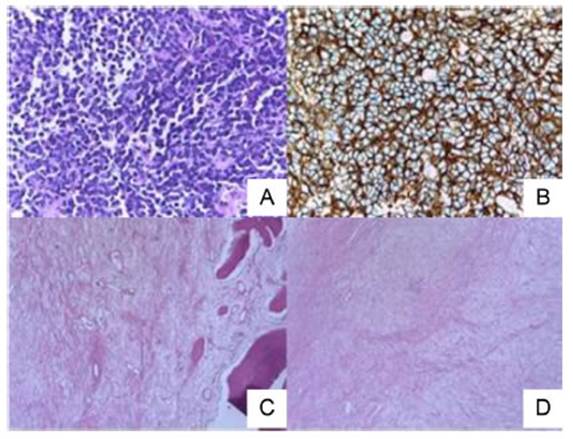
Fig. 2 Histopatología del tejido tumoral en material de biopsia pre-tratamiento (A y B) y pieza quirúrgica post-tratamiento (C y D). A: Tinción hematoxilina-eosina 40X. Se observa masa compacta de células pequeñas, redondas y uniformes con escaso citoplasma claro, divididas en lóbulos irregulares por hebras fibrosas. B: Tinción inmunohistoquímica 40X, positiva para CD99 de membrana plasmática de células tumorales (patrón membranoso) compatible con sarcoma de Ewing. C y D: Tinción hematoxilina-eosina 10X. Se observa remisión completa sin evidencia de tumor viable con áreas de fibrosis (C), hialinización (D) y presencia de macrófagos con hemosiderina en el área del tumor
Se inició tratamiento con quimioterapia según esquema VAC/IE (vincristina, doxorrubicina y ciclofosfamida alter nado con ifosfamida y etopósido); recibió 3 cursos de VAC y 2 cursos de IE con regular tolerancia, pero al desarrollar neurotoxicidad se redujo la dosis de vincristina. Concu rrente al tratamiento sistémico recibió radioterapia externa a dosis de 5040 cGys, en 28 sesiones. Un mes después, al examen clínico se observó tumor ubicado a nivel de 7mo-8vo arcos costales dorsales derecho con disminución del diámetro tumoral a 10 cm. La resonancia magnética de reevaluación mostró respuesta parcial al tratamiento y la biopsia fue negativa para neoplasia maligna.
Con estos hallazgos, el paciente fue sometido a resec ción del tumor y reconstrucción de la pared torácica con malla de polipropileno. En el acto operatorio se encontró una extensa tumoración de 16x10x9 cm dependiente del 7mo arco costal derecho con invasión al 9no arco costal en su aspecto posterior. En su extremo posterior se contactaba con las apófisis transversas derechas; para su extirpación, fue necesaria la resección parcial de una de ellas hasta el agujero de conjunción vertebral, sin invadirlo. La tumoración respetaba los tejidos blandos y la pleura parietal. En el estudio anatomopatológico de la pieza operatoria no se observó tumor viable, en su lugar se observaron áreas de fibrosis, hialinización y macrófagos con hemosiderina, con 5% de necrosis, 90% de fibrosis, 5% de degeneración quística y bordes quirúrgicos libres (Fig. 2 C y D). La evolución postoperatoria cursó sin com plicaciones y actualmente (6 meses post-tratamiento) el paciente se encuentra en seguimiento con tomografía de tórax negativa para recurrencia de enfermedad.
Discusión
El SE es un tumor maligno agresivo poco frecuente de hueso y tejidos blandos descrito por primera vez en 1921 por el patólogo James Ewing. Es uno de los clásicos tumo res de células pequeñas, redondas y azules, que ocurren predominantemente en la infancia y la adolescencia. La familia de los tumores de Ewing incluye el SE óseo, el tumor neuroectodérmico primitivo, el SE extraóseo y el tumor de Askin7. El 90% ocurre en la primera y segunda década de vida, y su aparición en mayores de 30 años es excepcional8. La localización es variable. En niños y adolescentes, la afectación del esqueleto axial representa el 54% (12% torácica) mientras que el esqueleto de las extremidades está comprometido en el 42% de los pa cientes7. Pocas series han comunicado la frecuencia de SE en adultos. Cesari y col. observaron que el 20% de los SE en pacientes mayores de 40 años se presentaron en la pared torácica y eran localmente avanzados, como nuestro caso, o metastásico8.
El SE de pared torácica se presenta como una masa sólida asociada o no a dolor y los estudios de imágenes más útiles son la tomografía de tórax y la resonancia magnética de pared torácica9. El diagnóstico anato mopatológico se basa en los hallazgos histológicos e inmunohistoquímicos. El marcador más útil es CD99, positivo en nuestro caso. Sin embargo, no es específi co, porque se expresa en una variedad de neoplasias mesenquimales10. Otros marcadores inespecíficos son S-100 proteína, neurofilamentos, citoqueratina y desmina, que fueron negativos en nuestro estudio. La inmunopositividad FLI-1 se puede observar en aquellos ES que albergan fusiones de genes EWSR1-FLI1, pero su ausencia, como ocurrió en nuestro caso, no descarta el diagnóstico de SE10. Para la confirmación definitiva, se utiliza el estudio de citogenética, la translocación entre los cromosomas 11 y 22, fusionando parte del gen EWSR1 y parte del gen FLI-17. En nuestro caso se confirmó mediante RT-PCR la presencia de genes de fusión EWS/FLI tipo 1. El diagnóstico diferencial incluye condrosarcoma mesenquimatoso, tumor des moplásico de células pequeñas y osteosarcoma de células pequeñas11.
El tratamiento de SE del adulto puede diferir del de los adolescentes debido a diferencias en la biología y el desarrollo tumoral8. Además, dada la rareza de la presen tación en adultos, no existen guías estandarizadas para su tratamiento y seguimiento, por lo que algunos autores han extrapolado los conocimientos del tratamiento en niños y adolescentes con resultados diversos8,12. Algunos estudios indican que la quimioterapia y el control local son esenciales para obtener resultados favorables en pacientes con SE localizado en pared torácica. Sham berger y col. establecieron la importancia de la quimiote rapia neoadyuvante y el tratamiento quirúrgico posterior, especialmente en pacientes cuyos tumores iniciales son de gran volumen3. Con respecto al control local de SE, aún es un tema de discusión si la radioterapia adyuvante beneficia la supervivencia. Seitz y col. informaron un mejor control local con radioterapia después de la ciru gía13. Otros autores solo la indican en aquellos casos en los que se presentan márgenes positivos después de la resección quirúrgica paliativa, cuando la resección com pleta es imposible3. En nuestro caso, el paciente recibió quimioradioterapia neoadyuvante, obteniendo respuesta patológica completa en la pieza operatoria la cual es un hallazgo raro, asociado a un buen pronóstico14. En 1998, Wunder y col. analizaron la respuesta histológica a la quimioterapia en 74 pacientes con SE, clasificándose de manera semicuantitativa en: grado I indicó una necrosis de < 50% del tumor; grado II, necrosis entre 50% y 90%; grado III, necrosis entre 90% y 99%; y el grado IV, necrosis de 100%. Se consideró que los grupos III y IV tuvieron una buena respuesta, con mejores tasas de superviven cia15. Albergo y col. encontraron que el pronóstico mejoró significativamente en quienes recibieron neoadyuvancia y alcanzaron 100% de necrosis en la pieza operatoria14.
Clásicamente se han descrito diferencias entre el SE de jóvenes y adultos. Estos últimos están asociados a peor pronóstico y mayor incidencia de tumores poco diferenciados15. Por otra parte, la revisión de Seker y col. no encontró diferencias estadísticas entre ambos grupos etarios; probablemente estos resultados inconsistentes se deban a bajo número de pacientes en el grupo de SE del adulto15. Se ha analizado el valor pronóstico de variables tales como edad, sexo, sitio del tumor primario, con resul tados ambiguos, salvo para la enfermedad metastásica que en sí misma es un factor de peor pronóstico9.
El tratamiento multimodal ha aumentado la tasa de su pervivencia en los últimos años. Jacobs y col. informaron una supervivencia global a 5 años de 71% en pacientes con SE de pared torácica no metastásico y de 49% en el grupo metastásico3,14. Además, Césari y col. observaron que la respuesta patológica completa se asoció a una sobrevida libre de enfermedad a los 5 años de 63%, porcentaje poco menor al obtenido en el tratamiento de niños y adolescentes8. Una supervivencia similar se logra con con terapia multimodal en adultos con SE8. En nuestro caso, 6 meses después de completar tratamiento quirúrgico no se evidenció recurrencia de enfermedad.
El SE comúnmente es considerado una enfermedad infantil. Rara vez se presenta en personas adultas y, cuando lo hace, por lo general, tiene peor pronóstico. El tratamiento multimodal de pacientes mayores de 40 años ha probado mejorar la respuesta patológica al tratamiento, la cual constituye un factor determinante de buen pronós tico y supervivencia.

