











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
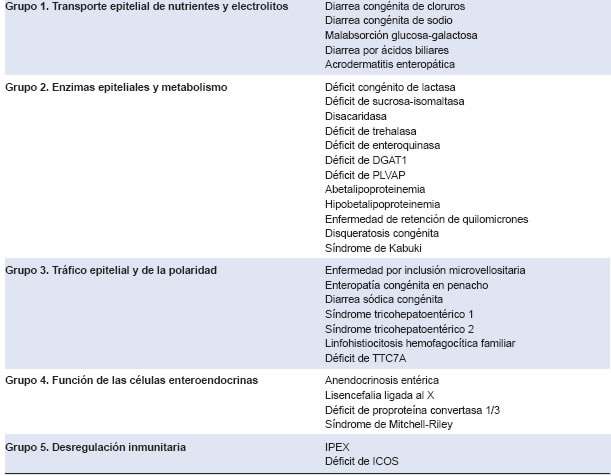
La diarrea es uno de los principales motivos de consulta en pediatría. En los últimos años, con el surgimiento de la secuenciación genómica, se ha identificado un nuevo grupo de diarreas crónicas: las diarreas y enteropatías congénitas (CODE por su sigla en inglés).1 Estas se clasifican en cinco grupos (Tabla 1). 1 En el grupo dos, podemos encontrar una enzima llamada diacilglicerol o-aciltransferasa 1 (DGAT1). La mutación del gen DGAT1 produce diarrea crónica grave, que se desarrolla principalmente en el período neonatal o en los dos meses posteriores al nacimiento.2
Este trabajo tiene como objetivo presentar a dos hermanas que consultaron por diarrea crónica y retraso del crecimiento, en quienes se diagnosticó una mutación en el gen que codifica DGAT1. Estas dos pacientes son las primeras encontradas con esta mutación en Latinoamérica.
CASO CLÍNICO 1
Niña recién nacida de término (RNT) de 38 semanas, con peso de nacimiento de 3410 g, talla de 50 cm, que consultó a los cuatro meses de vida por retraso en el crecimiento. No presentaba vómitos, pero, a pesar de que solo tenía una deposición al día, las heces eran muy líquidas. Se alimentaba con fórmula de inicio con aporte calórico adecuado, pero no aumentaba de peso. Al examen físico, presentaba desnutrición grave: peso de 4,340 kg (puntaje Z -2,9), talla de 59,5 cm (percentil 3, puntaje Z -2) y perímetro cefálico normal para la edad.
En el laboratorio inicial, presentaba anemia, trombocitosis, hipoalbuminemia e hipogammaglobulinemia.
Los estudios iniciales, que incluyeron ecografía abdominal, ecocardiograma, dos pruebas de sudor y estudio molecular para mutación en el gen regulador transmembrana de la fibrosis quística, no revelaron ningún hallazgo positivo. Las pruebas de materia fecal fueron negativas para virus, bacterias y parásitos; aclaramiento de alfa1-antitripsina aumentado, que sugería una enteropatía perdedora de proteínas.
Se realizó evaluación inmunológica y se evidenció disminución de la inmunoglobulina G con recuento de linfocitos normales (secundaria a la enteropatía perdedora de proteínas).
Debido a los hallazgos previos, se realizó una endoscopia digestiva alta y baja con biopsia, que mostró enteropatía grado II moderada, gastritis crónica inactiva leve y colitis inespecífica con eosinófilos elevados.
Debido a la falta de aumento de peso con fórmula extensamente hidrolizada, con sospecha de colitis eosinofílica, inició tratamiento con metilprednisolona a 2 mg/kg/día. Al no observarse respuesta, se realizó una segunda endoscopia digestiva, sugestiva de enteropatía en penacho congénita. Sin embargo, las pruebas moleculares para el gen EPCAM asociado con esta enfermedad no revelaron ninguna mutación.
La paciente recibió alimentación con fórmula extensamente hidrolizada, sin presentar aumento de peso a pesar de la ingesta calórica adecuada, por lo que inició nutrición parenteral. Permaneció internada durante un año con lenta recuperación nutricional y fue dada de alta con internación domiciliaria sin un diagnóstico claro.
CASO CLÍNICO 2
Dos años más tarde, consultó su hermana menor con síntomas similares. Era una RNT (38 semanas), con peso de nacimiento de 3200 g y talla de 49 cm. A los dos meses de vida, comenzó con vómitos, diarrea acuosa (una deposición al día) y retraso en el crecimiento. Al examen físico presentaba deshidratación leve y desnutrición grave: peso de ingreso de 3,580 kg (puntaje Z -3,3), talla de 56 cm (percentil 3-10) y perímetro cefálico normal para la edad.
Al igual que su hermana, presentaba anemia, trombocitosis, hipoalbuminemia e hipogammaglobulinemia.
Se realizaron los mismos exámenes complementarios, todos con resultados normales. Se realizó endoscopia digestiva alta y baja con resultados inespecíficos (enteropatía leve, gastritis crónica inactiva y mucosa colónica con edema).
La paciente no mostró aumento de peso a pesar de un aporte calórico adecuado con fórmula extensamente hidrolizada, la cual recibió desde el inicio de la internación, por lo que se inició nutrición parenteral.
Dado que la paciente presentaba los mismos síntomas que la hermana mayor, se solicitó un panel completo de diarrea congénita. El análisis mostró dos mutaciones heterocigóticas para el gen que codifica la enzima DGAT-1: c.838C>T (p.Arg280*) clasificada como patógena y c.1162C>T (p.His388Tyr) clasificada como probablemente patógena según las normas del Colegio Americano de Genética Médica y Genómica.
Sobre la base de estos hallazgos, se realizó la secuenciación directa de los exomas 9 y 15 del gen DGAT1 de ambos padres y de la hermana mayor, con los siguientes resultados:
Madre: variante c.1162C>T (p.His388Tyr) en estado heterocigoto y ausencia de la variante c.838C>T (p.Arg280*).
Padre: variante c.838C>T (p.Arg280*) en estado heterocigoto y ausencia de la variante c.1162C>T (p.His388Tyr).
Hermana mayor: tanto la variante p.ARg280* como la p.His388Tyr se encontraron en estado heterocigoto.
Con estos hallazgos, se realizó el diagnóstico de diarrea congénita secundaria a déficit de DGAT1 y se brindó asesoramiento genético a los padres. Como resultado, ambas niñas recibieron una dieta restringida en grasas (carbohidratos, como por ejemplo arroz, carne magra y verduras), y mejoraron su estado nutricional.
Posteriormente se suspendió la nutrición parenteral en ambas pacientes y mantuvieron adecuada ganancia de peso. La hermana mayor tiene actualmente tres años y siete meses, presenta un peso de 15,1 kg (percentil 25-50), talla de 100,5 cm (percentil 70) y peso para la talla del 92 %; mientras que la hermana menor tiene un año y seis meses, peso de 9,530 kg (percentil 50-75), talla de 80,5 cm (percentil 25 50) y peso para la talla del 92,6 % (Figura 1).
DISCUSIÓN
Las CODE son causas raras pero graves de diarrea crónica en los niños. En los últimos años, debido a los avances en la secuenciación genómica, se han identificado muchos defectos monogénicos, incluido el déficit de DGAT1. El gen reside en el cromosoma 8 humano y codifica para la enzima DGAT. Esta es responsable de catalizar el paso final en la síntesis de triglicéridos, usando diacilglicerol y acil-graso CoA.3 Aunque varios estudios sugieren que el déficit de DGAT1 aumenta la sensibilidad a la toxicidad inducida por lípidos y apoptosis de las células epiteliales intestinales dando como resultado enteropatía con pérdida de proteínas,4,5 la causa de la diarrea sigue siendo desconocida.
Actualmente, no se ha identificado una correlación genotipo-fenotipo, ya que el resultado varía desde la resolución completa de los síntomas gastrointestinales hasta un curso letal de la enfermedad.6 Los pacientes con déficit de DGAT1 pueden presentar distintas manifestaciones clínicas: enteropatía perdedora de proteínas, vómitos y/o diarrea, intolerancia a las grasas y retraso en el crecimiento.4 Además, según reportes anteriores, puede causar una diarrea de aparición temprana, crónica e intratable, que lleve a la insuficiencia intestinal.2
El vómito es otro síntoma importante, reportado en la mayoría de los pacientes. Una explicación probable para tal síntoma sería que el déficit de DGAT1 inhibe la secreción de quilomicrones y retrasa el vaciamiento gástrico.7
Como se describió anteriormente, nuestras pacientes tenían hipoalbuminemia e hipogammaglobulinemia; sin embargo, a diferencia de reportes previos, los niveles de triglicéridos fueron normales en ambos casos.
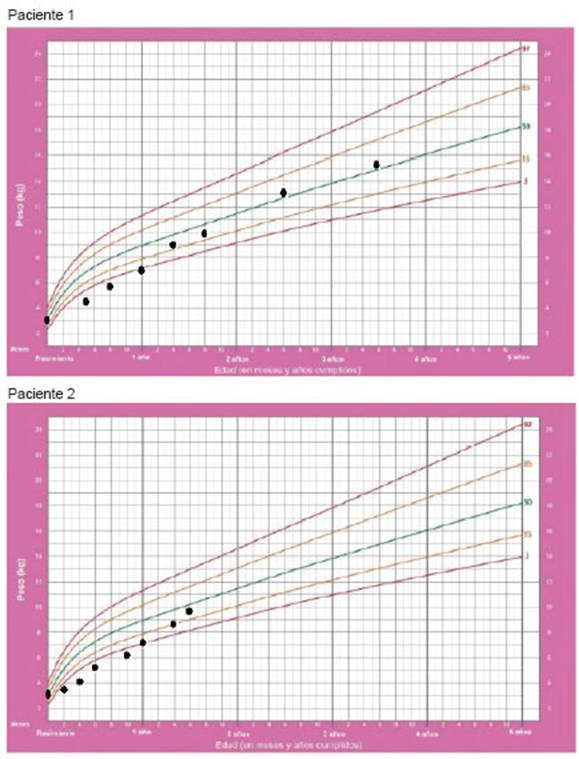
Figura 1: Gráfico de peso para la edad para niñas desde el nacimiento hasta los 5 años. Estándares de crecimiento infantil de la Organización Mundial de la Salud (OMS)
Se han descrito diferentes mutaciones en el gen DGAT1, la mayoría en estado homocigota y en individuos de ascendencia asiática o judía asquenazí. En nuestras pacientes se encontraron dos variantes en diferentes regiones codificantes (exones 5 y 9), una de las cuales no había sido reportada previamente.
La primera variante, NM_012079.6:c.838C>T, da como resultado un codón de parada que produce pérdida de función, un mecanismo conocido de enfermedad para este gen. Esta variante fue reportada como una mutación causante de enfermedad en ClinVar (última consulta realizada el 19 de agosto de 2021).
La segunda variante, c.1162C>T en DGAT1, es una mutación de sentido erróneo y produce un cambio de aminoácido en la región MBOAT donde se han informado previamente otras mutaciones. Aunque esta mutación no se ha encontrado en otros pacientes, estudios clínicos de correlación y segregación avalan su patogenicidad.
El tratamiento de esta enfermedad consiste en una dieta restringida en grasas, donde la administración de ácidos grasos de cadena corta demuestra ser un buen complemento en la dieta de algunos pacientes.6 Ambas pacientes mostraron una excelente mejoría después del inicio de la dieta.
CONCLUSIÓN
En este trabajo presentamos dos hermanas con mutaciones del gen DGAT1 heterocigotas que se manifestaron con retraso en el crecimiento y diarrea. Hasta donde sabemos, estos son los dos primeros pacientes con este diagnóstico en Latinoamérica. Estos casos nos permiten ampliar nuestro conocimiento sobre las diarreas congénitas en general y sobre el déficit de DGAT1 en particular. Debido al curso grave que esta enfermedad puede tener, los pediatras deben considerar esta condición en lactantes pequeños que se presentan con diarrea crónica y retraso en el crecimiento, luego de que otras causas más frecuentes hayan sido descartadas.

