











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La leucemia cutis es una patología infrecuente y se puede asociar a diferentes tipos de leucemia. En la leucemia mieloblástica aguda (LMA), un 30 % de los casos presentan manifestaciones extramedulares asociadas con mayor frecuencia a la variante monoblástica (FAB M5) con o sin compromiso medular asociado.1 La incidencia es mayor en la población neonatal y durante el primer año de vida: esta forma de presentación asciende hasta un 50 % y suele asociarse a síndromes genéticos, como las trisomías 21,13 y 9, y el síndrome de Bloom.2 Las lesiones cutáneas aparecen con mayor frecuencia después del diagnóstico hematológico, son concomitantes hasta en un 30 % y anteriores en menos del 10 %.3,4
Se desconoce la causa por la que se da la migración de las células leucémicas a la piel. Una de las hipótesis sostiene que hay migración impulsada por el receptor 4 de citocinas, el cual presenta una sobreexpresión aberrante en los blastos, atrayéndolos hacia este sitio.3 Es importante tener en cuenta que las leucemias de los pacientes menores de un año frecuentemente presentan hiperleucocitosis y compromiso extramedular que se manifiesta como organomegalias, afectación de piel y/o sistema nervioso central (SNC).5 La presencia de compromiso de piel no influye en el pronóstico de la enfermedad, que es desfavorable; alcanza según las series más representativas tasas de sobrevida del 20-30 %.67 Además, se describe que, cuando se observa compromiso cutáneo, la incidencia de la afectación a nivel del SNC es frecuente, como expresión de diseminación de la enfermedad.3,7,8
Los diagnósticos como infecciones, otras patologías neoplásicas con afectación cutánea y los trastornos histiocíticos, entre otros, constituyen los principales diagnósticos diferenciales ya que configuran un escenario pronóstico y terapéutico diferente.
CASO CLÍNICO
N.° 1Paciente varón de 5 meses sin antecedentes de relevancia que consultó en su hospital de origen por múltiples pápulas y tumores de consistencia duro-elástica de 1-2 cm de diámetro, rosados, homogéneos, distribuidos en cuero cabelludo, cara, dorso y área del pañal, no dolorosos, de 2 meses de evolución (Figura 1).
Se solicitó hemograma, que presentó recuento de leucocitos normal con predominio linfocitario. Se realizó biopsia de una lesión dorsal, que informó infiltración mononuclear dérmica, con Ki67 30-40 % e inmunohistoquímica (IHQ) diagnóstica de proliferación mielo-mononuclear. La citometría de flujo fue positiva para HLA-DR, CD14, CD64, CD13 y CD11b, marcadores mielo-monocíticos.
Con sospecha inicial de leucemia cutis, consultó en nuestra institución, donde se realizó punción aspiración de médula ósea (MO) y se descartó infiltración leucémica, con análisis de transcripción reversa seguida de reacción en cadena de la polimerasa (RT-PCR) para descartar alteraciones del gen KMT2A, frecuentes en pacientes menores de 1 año con leucemia aguda, que resultaron negativos. Se realizó revisión del taco de anatomía patológica y nueva biopsia de piel, que mostró dermis con histiocitos xantomizados con células gigantes multinucleadas, e IHQ positiva para CD68 en los histiocitos; se confirmó el diagnóstico de xantogranuloma juvenil diseminado (XJD) (Figura 2).
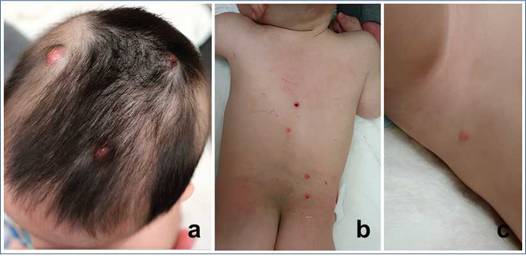
Figura 1: Fotografías del caso número 1. Se pueden observar pápulas y tumores eritemato-anaranjadas en la región cefálica (a), tronco (b) y axila (c)
Se realizaron estudios de estadificación y se encontró compromiso renal, pulmonar y hepático como manifestaciones de la forma diseminada de la enfermedad. El paciente inició tratamiento según el protocolo colaborativo internacional de la Histiocyte Society (LCH-IV). Actualmente, se encuentra en mejoría pero con enfermedad activa.
CASO CLÍNICO N.° 2
Paciente varón de 11 meses con antecedente de dermatitis atópica, que consultó por fiebre y neutropenia asociadas a nódulos subcutáneos rosados, no dolorosos, de 1 cm de diámetro, localizados en región cefálica y miembros superiores, de 5 meses de evolución.
Previamente, en otro centro se realizó biopsia de una lesión que informó leucemia cutis. Se realizó revisión del taco que mostró proliferación mieloide dermo-hipodérmica, con expresión de lisozima, positividad intensa para CD68 y CD43 focal (Figura 3). Se agregaron determinaciones IHQ, se encontró HLA DR y CD34 negativos, lo cual desestimó el diagnóstico de leucemia, que también fue descartado por citometría de flujo.
Se realizaron simultáneamente punciones de MO, cuyo informe histológico mostró aumento de la trama reticulínica, hemofagocitosis y freno madurativo mieloide, sin células neoplásicas. Se descartaron alteraciones moleculares asociadas a leucemia por RT-PCR.
Dada la disminución de precursores mieloides, se realizó el estudio de fallos medulares y se encontró déficit de hierro y vitaminas D y B12. Se realizó secuenciación del gen ELANE y SDBS, sin encontrarse alteraciones génicas con implicancia fenotípica; estudio de longitud telomérica que fue normal, test de diepoxibutano negativo y Next Generation Sequencing (NGS) que no mostró variantes de ADN compatibles con el cuadro del paciente. También se descartaron inmunodeficiencias.
El niño recibió factor estimulante de colonias (GM-CSF) con buena respuesta. Actualmente, no presenta neutropenia y las lesiones cutáneas se encuentran resueltas.
DISCUSIÓN
Se presentaron dos casos en los cuales la hipótesis diagnóstica inicial fue de leucemia cutis, patología infrecuente que se asocia en este grupo etario a LMA y en la cual el 3050 % presenta manifestaciones extramedulares asociadas a la variante FAB M5.1-4 Ambos pacientes presentaban edades en el rango de mayor incidencia de esta patología, sin embargo, es importante tener en cuenta que las leucemias en lactantes suelen presentarse con hiperleucocitosis, asociadas o no a leucostasis y compromiso extramedular, que se manifiesta por la infiltración de hígado, bazo, piel y SNC.5-7 Al examen histopatológico se presenta un infiltrado denso de células inmaduras dentro de acúmulos Dadas estas características, es probable que se presente coagulación intravascular (CID), que afecta el estado general.8
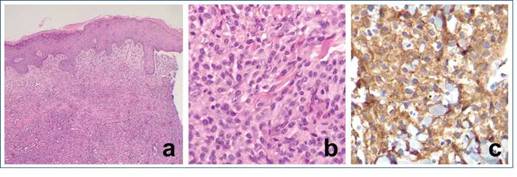
Figura 2: Anatomía patológica del caso número 1. En (a) se observa una proliferación celular densa en dermis reticular (hematoxilina-eosina, objetivo de campo). En (b) se observan células de mediano tamaño, con citoplasma eosinófilo con finas vacuolas (hematoxilina-eosina, objetivo 40X) que son positivas para CD163 (c), que evidencian la naturaleza histiocítica de la lesión de colágeno en la dermis reticular y vasculitis.
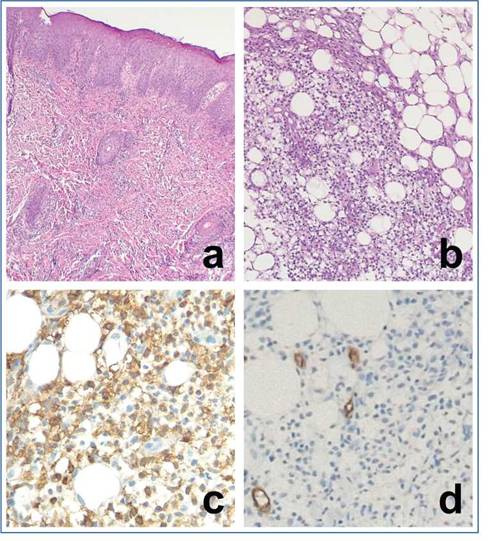
Figura 3: Anatomía patológica del caso número 2. Se puede observar en (a) y en (b) piel con infiltración celular densa, más evidente en hipodermis que incluye linfocitos maduros y en maduración (hematoxilina eosina, objetivo de campo). En (c) se observa que las células infiltrativas son positivas para mieloperoxidasa, mientras que son negativas para CD34 (d). La marcación positiva en (d) corresponde al endotelio vascular
En los casos presentados, el estado clínico de ambos pacientes era bueno, no presentaban recuento leucocitario elevado, organomegalias, ni signos de CID.
La presencia de compromiso cutáneo no influye en el pronóstico de la leucemia, que es desfavorable; alcanza según las series más representativas tasas de supervivencia del 20-30 %.6,7 Además, cuando se observa compromiso cutáneo, la incidencia de la afectación a nivel del SNC es frecuente.7,8 El método estándar de oro para el diagnóstico es la biopsia de las lesiones con IHQ y estudio de biología molecular para detectar reordenamientos del gen KMT2A (previamente llamado gen MLL) el cual transloca con el gen MLLT3 generando el transcripto de fusión KMT2A-MLLT3/t(9;11)(p21.3;q23.3), y se asocia a LMA M5 de novo en menores de un año, lo que configura un pronóstico desfavorable, con una mortalidad del 80 % al año del diagnóstico.7,8
Las biopsias de ambos pacientes presentaron infiltrados mononucleares con IHQ, CFM y estudio de biología molecular negativos.
Es importante tener en cuenta patologías que presentan similitud clínica con la leucemia cutis: en el caso 1, el diagnóstico fue XGJ, patología histiocitaria que corresponde al grupo “C” de histiocitosis (solo afectación cutánea) o al grupo “L” (afectación de otros órganos). Las características histológicas son idénticas: se observan histiocitos xantomizados y células gigantes tipo Touton y tipo cuerpo extraño. El origen de estas células es desconocido, aunque se postula que comparten características con los macrófagos dérmicos, ya que expresan factor XIIIa. Con IHQ se observa que los histiocitos expresan CD68 (100 %), vimentina, factor XIIIa (99 %) y lisozima, y son negativos para CD34, actina muscular lisa, S-100, CD1a y langerina. Los hallazgos moleculares encontrados en pacientes pediátricos incluyen mutaciones activantes de la vía MAPcinasa (proteincinasas activadas por mitógenos) (BRAF V600), fusiones de NTRK 1 y mutaciones en MAP2K1 y CSF1R9.
Clínicamente, el XGJ se presenta como una lesión pápulo-nodular única o múltiple, de color rosado o amarillento, asintomática, localizada en cualquier región corporal en pacientes de entre 6 meses y 2 años.10 La forma diseminada de la enfermedad es excepcional; la cámara anterior ocular es la localización extracutánea más frecuente; sigue en frecuencia pulmón, hígado, meninges, bazo y testículo.11 Se trata de una patología benigna y, en general, no requiere tratamiento, sin embargo, si se presenta compromiso sistémico, se puede utilizar un esquema de corticoides y vinblastina.12
En el caso 2 el paciente presentó neutropenia asociada. Esto motivó inicialmente la búsqueda de causas secundarias, como infecciones, principalmente del grupo TORCH, que se manifiestan con hepatoesplenomegalia, anemia, trombocitopenia y el aspecto blueberry muffin syndrome con nódulos subcutáneos azulados.13 Otro diagnóstico diferencial en lactantes con compromiso cutáneo y del hemograma es el neuroblastoma, tumor maligno que desde la presentación congénita hasta los 18 meses puede implicar compromiso cutáneo en un 2 % y asocia compromiso de MO.14 Sin embargo, otra característica importante en esta patología es el compromiso hepático por infiltración, situación que genera frecuentemente dificultad respiratoria restrictiva y condiciona el estado general del paciente, que en este caso era bueno.
Por último, en relación con el compromiso cutáneo en errores innatos de la inmunidad, se describe de forma infrecuente la presencia de nódulos fríos asociados a la inmunodeficiencia común variable, la grave combinada y los síndromes granulomatosos crónicos.15 En este niño, el antecedente de dermatitis atópica y neutropenia obligaron a descartar esta entidad. Además, se realizó el estudio del fallo medular mieloide con estudios de complejidad creciente, desde la búsqueda de malabsorción, intoxicaciones e inmunodeficiencias, hasta la secuenciación genómica.
En ambos pacientes fue fundamental el abordaje interdisciplinario para llegar al diagnóstico correcto, el cual en ambos casos configuró escenarios diferentes en cuanto a tratamiento y pronóstico.
Recibido: 27-7-2022
Aceptado: 20-12-2022

