










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Ciencia y Tecnología
On-line version ISSN 1851-7587
Rev. cienc. tecnol. no.12 Posadas July/Dec. 2009
GENÉTICA - BIOLOGÍA
Optimización de la amplificación mediante PCR de la región promotora 1 del gen shp-1 luego de su tratamiento con bisulfito
Amplification optimization through PCR for region 1 promoter of gen shp-1 in bisulfite treated region
E. Martín Giorgio1, Bárbara J. Chaneton1, Pedro D. Zapata1
1-Cátedra de Biología Celular y Molecular (Bioquímica), Módulo de Bioquímica y Farmacia, Facultad de Ciencias Exactas, Químicas y Naturales, UNaM. Posadas, Misiones, Argentina. Av. Mariano Moreno 1375. Posadas, Misiones, Argentina (3300). bcmb@fceqyn.unam.edu.ar.
• Martín E. Giorgio1 Es alumno avanzado de la Licenciatura en Genética. Se desempeña actualmente como Investigador Auxiliar en el proyecto acreditado 16Q361. Ha participado en calidad de tesista del desarrollo del proyecto subsidio de la ANPCyT (PICT 05–15058) y participa también de otros proyectos relacionados con desarrollos biotecnológicos. Es Auxiliar de Segunda en las Cátedras de Biología Celular y Molecular (Bioquímica) y Biotecnología Molecular (Ingeniería Química, Bioquímica y Farmacia). Posee 4 publicaciones en revistas indexadas.
• Bárbara J. Chaneton1 Es alumno de 4º año de la Licenciatura en Genética. Se desempeña actualmente como Auxiliar en el proyecto acreditado 16Q361. En el marco de este proyecto posee 1 publicacion en la revista BAG y ha presentado trabajos en las Jornadas de Ciencia y Tecnología de la FCEQyN.
• Pedro D. Zapata1 Es Doctor por la Universidad de Alcalá de Henares, en el programa Biomedicina. Se desempeña actualmente como Profesor Regular Adjunto en las Cátedras de Biología Celular y Molecular (Bioquímica), Genética Molecular (Lic. en Genética), Biología Celular (Lic. en Genética) y Biotecnología Molecular (Ingeniería Química, Bioquímica y Farmacia). Posee actualmente la Categoría III en el Sistema Nacional de Incentivos a los Docentes–Investigadores. En el área de Biomedicina ha sido beneficiario de un subsidio de la ANPCyT (PICT 05–15058) y actualmente dirige proyectos acreditados en el CIDET. Posee 37 publicaciones en revistas indexadas y 25 presentaciones a congresos.
Resumen
Las alteraciones en los patrones de metilación de algunos genes pueden contribuir al desarrollo del cáncer. Existen evidencias de cambios en los niveles de expresión de SHP-1 en próstata tumoral, siendo fundamental evaluar el estado de metilación de sus promotores. La secuenciación bisulfito es la técnica de elección para establecer el patrón de metilación. El objetivo de este trabajo fue estandarizar las condiciones para la amplificación del Promotor 1 para el gen shp-1 luego del tratamiento con bisulfito. Se trabajó con 2 tejidos de expresión diferencial para SHP-1: mucosa yugal y tejido sanguíneo. A partir de cebadores que hibridan en sitios libres de islas CpG del promotor 1 del gen shp-1 tratado con bisulfito de sodio se analizaron distintas combinaciones de los componentes de PCR aplicándose distintas estrategias de ciclado. Un esquema de ciclado con 3 bloques de temperatura de hibridación decreciente permitió la obtención del amplicón de 500pb coincidente con el esperado para el fragmento del promotor analizado.
Palabras Clave: Epigenoma; Tratamiento con bisulfito; PCR; Shp-1.
Abstract
Changes in methylation patterns may contribute to cancer development. Recent studies have shown that shp-1 expression is diminished in prostate cancer. Accordingly, it is relevant to evaluate the methylation pattern of its promoters. Bisulfite sequencing is the reference technique for these studies. The aim of this paper was the standardization of the conditions for shp-1 gene Promoter 1 amplification before bisulfite treatment. Two tissues with differential expression of shp-1 were investigated: buccal epithelial cells and blood. We analyzed different combinations of PCR component with primers that hybridized in CpG island free zones. The application of 3 segments of decreased hybridization temperature allowed us to obtain 500pb predicted amplicons for this fragment of promoter 1.
Key Words: Epygenome; Bisulfite treatment; PCR; Shp-1
Introducción
La regulación epigenética incluye una serie de procesos que modifican la expresión génica pero que no conllevan modificaciones de la secuencia de DNA [1]. Estos cambios son heredables en el patrón de expresión de un gen de una célula a otra, aunque no de un individuo a otro [2]. Estas modificaciones topológicas del DNA incluyen adición covalente de grupos funcionales como metilos, acetilos y fosfatos, unión de proteínas al DNA y modificación de proteínas asociadas al DNA.
Los cambios epigenéticos tienen repercusiones en la regulación transcripcional modificando la expresión de genes, pudiendo contribuir al desarrollo de patologías como el cáncer, la esquizofrenia y el asma [1]. Muchos de estos cambios incluyen la metilación del DNA en regiones promotoras de genes, siendo una consecuencia importante la inactivación de genes supresores de tumores [2, 3].
La metilación es uno de los parámetros que regula la transcripción, estando asociada la hipermetilación del promotor con la ausencia de transcripción. Sin embargo, es uno de entre varios eventos reguladores que influyen en la expresión génica y es aplicable a ambas copias alélicas de un gen, aunque en ocasiones puede distinguir alelos maternos y paternos lo que daría diferencias en su expresión. [4]
La metilación suele ocurrir en el carbono 5 de las C precedidas por una G (islas CpG) y es catalizada por DNA metiltransferasas utilizando S–adenosil–metionina como dador de grupos metilos [2, 5]. Esta distribución de dinucleótidos CpG en el genoma es particular y el proceso de metilación podría ser necesario, pero no suficiente, para regular la trascripción de un gen [4].
En la actualidad existen pruebas que demuestran la importancia del control epigenético en el desarrollo del cáncer, observándose alteraciones en los patrones de metilación de genes relacionados con procesos de diferenciación, control del ciclo celular y adhesión [6–10].
Existen varias estrategias experimentales para evaluar la metilación de distintas zonas del genoma, siendo de especial interés las regiones promotoras [5]. Dos de las técnicas más utilizadas son la digestión con enzimas de restricción sensibles a metilación y la PCR metilación específica (MS–PCR). Sin embargo, en algunos casos el perfil de hipermetilación de las regiones promotoras es característico para cada tipo de cáncer, mientras que en otros se han observado perfiles similares o asociados a algún tipo de tejido [9]. En este sentido, las metodologías citadas permiten tener una visión parcial del estado de metilación, siendo conveniente la secuenciación del genoma tratado con bisulfito de sodio la técnica más sensible y específica para establecer el patrón de metilación de determinadas regiones génicas.
Nephew y Huang [10] proponen un modelo que intenta explicar la influencia de la metilación en la iniciación y progresión del cáncer. Normalmente existen algunas islas CpG que se encuentran protegidas contra la metilación aberrante, este modelo propone la pérdida de esta protección y la metilación de novo ocurriría progresivamente en los sitios flanqueantes a estas islas, lo que resultaría en el silenciamiento del gen correspondiente. Esta metilación se expandiría luego a otros loci importantes en estados más avanzados de un cáncer aumentando la densidad de metilación. De esta manera se plantea un progresivo cambio en el patrón de metilación durante el avance del tumor que tendría, como consecuencia, una alteración en el patrón de expresión génico. Por esta razón es importante establecer el patrón completo de metilación de una región génica.
A pesar de la relevancia del estudio de las regiones metiladas en muchos procesos patológicos, tales como el cáncer, en la literatura no existen muchas publicaciones que reparen en las cuestiones metodológicas implicadas en la puesta a punto del proceso de metilación, de manera que permita su posterior replicación. Teniendo en cuenta lo anterior, el objetivo de este trabajo fue estandarizar las condiciones metodológicas para la amplificación de una región del Promotor1 del gen shp–1 luego de su tratamiento con bisulfito para posteriormente iniciar el proceso de clonación de la misma y su secuenciación con fines de caracterizar su estado de metilación en diferentes tejidos.
Materiales y Métodos
Materiales
Dado que el objetivo del trabajo es la estandarización del método de amplificación de una porción del Promotor 1 para SHP–1 se trabajó con 2 tejidos de expresión diferencial para esta proteína: mucosa yugal y tejido sanguíneo.
Extracción de DNA
Para la extracción de ácidos nucleicos se utilizó la modificación de la técnica de salting out descripta por Miller et al [10] agregando un paso adicional de purificación con acetato de potasio. El DNA obtenido fue resuspendido en agua estéril libre de nucleasas y posteriormente fue almacenado a –20 ºC hasta su utilización.
Modificación química de citosinas
El tratamiento del DNA con bisulfito de sodio provoca la desaminación de las citosinas no metiladas, generando uracilo. Esto se realizó según el protocolo descripto por Herman et al [5] con una modificación en la metodología de purificación, que en este caso se realizó con el sistema DNA PURIFICATION KIT #K0513 (Fermentas). Para detener la desulfonación se adicionó NaOH 3 M. El DNA fue precipitado con 0,5 mL de isopropanol durante toda la noche a 4 ºC, se centrifugó a 12000 rpm por 5 min, se lavó con etanol 70 % v/v y se resuspendió en agua estéril libre de nucleasas, almacenándoselo a –20 ºC hasta el momento de su utilización.
Diseño bioinformático de cebadores
Para el diseño de los cebadores se utilizó la secuencia de GeneBank AB079851. Los cebadores fueron diseñados manualmente utilizando las secuencias modificadas en todas las citosinas y posteriormente analizados mediante los programas para diseño y análisis de cebadores Vector NTI 10.3.0 de Invitrogen Corporation y FastPCR 3.6.68 (Ruslan Kalendar, Institute of Biotechnology, University of Helsinki, Finland).
Amplificación por PCR
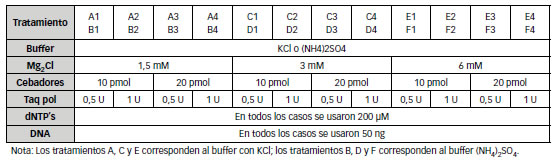
Las amplificaciones mediante PCR se realizaron utilizando buffer de PCR 1X conteniendo KCl o (NH4)2SO4, 200 μM dNTPs, y cantidades variables de cada cebador (OPERON Biotechnologies), MgCl2 y Taq polimerasa (Fermentas). En todos los casos se utilizaron 50 ng de DNA. Para cada una de las combinaciones resultantes se aplicaron distintas estrategias de ciclado consistentes en amplificaciones de T decreciente tipo touch down (Sambrook & Russell, 2001). (Tabla 1)
Tabla 1: Combinaciones analizadas en la mezcla de PCR para la amplificación del promotor metilado
Los productos de amplificación fueron separados en geles de agarosa al 2 % p/v, teñidos con bromuro de etidio 10 mg/ml, visualizados bajo luz ultravioleta y fotografiados con cámara digital Acer CS–5531.
En los casos en los que se logró el perfil de amplificación deseado se realizaron geles desnaturalizantes de poliacrilamida al 12 %. Los geles fueron teñidos con nitrato de plata y digitalizados mediante HP Desjekt F380. De estos geles se recortaron las bandas de tamaño adecuado y se resuspendieron en agua libre de DNasas para su posterior clonación y secuenciación.
Resultados y Discusión
La secuenciación bisulfito es el método de referencia para verificar la presencia de cambios puntuales en el patrón de metilación, sin embargo un punto crucial de esta metodología es la estandarización de la amplificación del promotor tratado con bisulfito. Habitualmente el tratamiento produce una gran cantidad de fragmentos de DNA por la agresividad química del mismo. Simultáneamente se produce un desequilibrio en el contenido de GC lo que genera regiones ricas en A y T, con la consecuente disminución de la especificidad de las secuencias. Todos estos inconvenientes repercuten en problemas de amplificación, siendo necesaria una adecuada estandarización de las condiciones de PCR. Esto implica el análisis pormenorizado del proceso paso a paso, detallándose a continuación los resultados para cada uno:
Diseño de cebadores
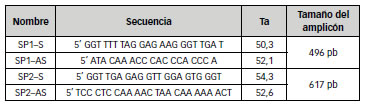
Con el fin de amplificar todo el promotor de manera independiente de su metilación se diseñaron 4 cebadores, 2 sentidos y 2 antisentidos que hibridarían en el promotor luego del tratamiento con bisulfito abarcándose un total de 36 sitios CG que hipotéticamente podrían presentar cambios en su estado de metilación. El par denominado SP1 amplificaría la secuencia más alejada del punto de origen de la replicación entre -847 y -351, mientras que el par denominado SP2 amplificaría la secuencia más próxima incluyendo el nucleótido +1 (-403 a +214). En ambos casos se eligieron zonas libres de islas CpG (Figura 1) y se analizaron las características termodinámicas de los cebadores a fin de evitar la formación de dímeros y la autocomplementariedad (Tabla 2). El análisis bioinformático no evidenció problemas de autocomplementariedad del los cebadores ni formación de dímeros.
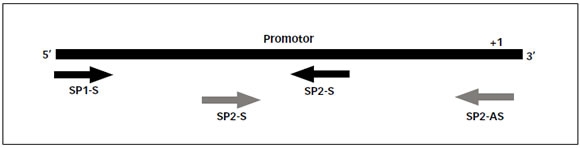
FIGURA 1. Estrategia utilizada para el diseño de cebadores. Referencias: SP1-S: cebador sentido para el fragmento SP1; SP1-AS: cebador antisentido para el fragmento SP1; SP2-S: cebador sentido para el fragmento SP2; SP2-AS: cebador antisentido para el fragmento SP2.
Tabla 2: Características de los cebadores diseñados
Estandarización de la PCR
Todos los componentes de la mezcla de amplificación representan variables a ser consideradas al estandarizar una PCR. Sin embargo, algunas de las variables pueden mantenerse constantes dado su menor influencia. En este trabajo se planificaron cambios en el tipo de tampón utilizado y en las concentraciones de Mg2Cl, cebadores y polimerasa, diseñándose una combinatoria de reacciones (Tabla 1) obteniéndose los mejores resultados con el tratamiento C3. El esquema de ciclado fue otro de los parámetros evaluados, analizándose distintas estrategias, resumidas en la Tabla 3.
Tabla 3: Estrategias de ciclado utilizadas
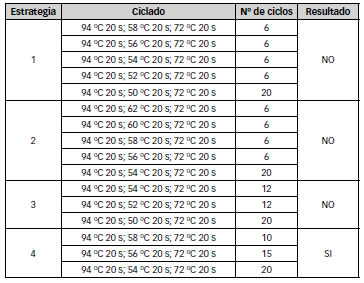
Únicamente se logró obtener el amplicón buscado de 500 pb para SP1 con esquemas de ciclado consistentes en 3 segmentos de distintas temperaturas de anneling: 58 ºC, 56 ºC y 54 ºC, habiéndose realizado diferentes variaciones en el número de ciclos de cada segmento ( Figura 2).
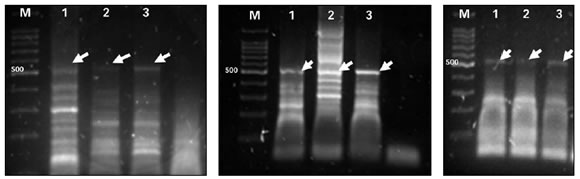
FIGURA 2. Amplificación mediante el tratamiento C3 del fragmento SP1 con un esquema de ciclado consistente en 94 ºC por 20 s, Ta (temperatura de anneling) variable por 20 s, 72 ºC por 20 s; siendo la Ta en cada caso de: A) 58 ºC (10 ciclos), 56 ºC (15 ciclos) y 54 ºC (20 ciclos). B) 58 ºC (20 ciclos), 56 ºC (12 ciclos) y 54 ºC (12 ciclos). C) 58 ºC (12 ciclos), 56 ºC (12 ciclos) y 54 ºC (20 ciclos). M: marcador de peso molecular; 1 y 2: sangre; 3: mucosa. C: control negativo
Como puede observarse, en ningún caso se obtuvo un único amplicón, pudiendo ser esto consecuencia de la inespecificidad de las regiones de hibridación luego del tratamiento con bisulfito. Teniendo en cuenta estos resultados, se seleccionó el ciclado que permitió obtener el amplicón de 500 pb con la menor cantidad de otros amplicones inespecíficos, es decir segmentos de temperatura de hibridación decreciente con 12 ciclos a 58 ºC, 12 ciclos a 56 ºC y 20 ciclos a 54 ºC.
Considerando que este protocolo de amplificación será utilizado para obtener amplicones para luego purificarlos, clonarlos y secuenciarlos, se separaron 10 muestras tratadas con bisulfito y amplificadas en geles de poliacrilamida (Figura 3), se recortaron las bandas y se reamplificó el producto obtenido para certificar la única presencia del amplicón deseado (Figura 4). Los resultados positivos obtenidos con estos dos últimos experimentos demuestran la utilidad de la estrategia planteada al poner a punto la amplificación.
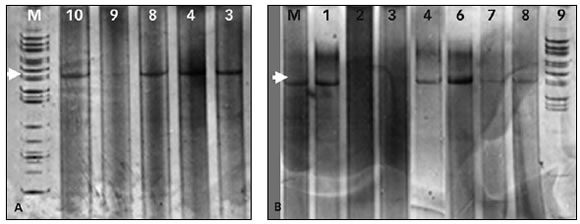
FIGURA 3. Gel de poliacrilamida desnaturalizante al 12%. Las muestras de DNA extraído a partir de sangre fueron amplificadas mediante el tratamiento C3 del fragmento SP1 con un esquema de ciclado de 58ºC (12 ciclos), 56ºC (12 ciclos) y 54ºC (20 ciclos). Se indica el marcador de peso molecular (M) y los números correspondientes a cada una de las muestras. A y B corresponden a dos experimentos distintos.
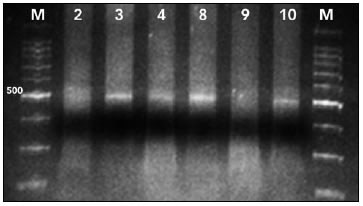
FIGURA 4. Reamplificación mediante tratamiento C3 de los amplicones obtenidos a partir del PAGE (Figura 5).
Conclusiones
Se diseñaron dos pares de cebadores (SP1 y SP2) que hibridan en sitios libres de islas CpG del promotor 1 del gen shp–1 tratado con bisulfito de sodio.
La utilización de un esquema de ciclado con 3 bloques de temperatura de hibridación decreciente con la mezcla de PCR C3 permitió la obtención de un amplicón de 500 pb coincidente con el esperado para el fragmento SP1 del promotor metilado y permitió su aislamiento a partir de geles de poliacrilamida y posterior reamplificación.
1. NEISS A J., GUERRERO C. Actualización del Código Genético al Código Epigenético: nuevas estrategias terapéuticas. Rev. Fac. Med. Univ. Nac. Coloma, 52(4): p. 287–230. 2004. [ Links ]
2. HERMAN G., BAYLIN M. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N. Engl. Med., 349: p. 2042–2054. 2003. [ Links ]
3. BAYLIN S. Mechanisms underlying epigenetically mediated gene silencin in cancer. Cancer Biology, 12: p. 331–337. 2002. [ Links ]
4. HSIEH CH . Dynamics of AND methylation pattern. Genetics & Development, 10: p. 224–228. 2000. [ Links ]
5. Herman J., Graff J., Myohanen S., Nelkin B., Baylin S. Methylation– Specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA. 93: p. 9821–9826. 1996. [ Links ]
6. MELKI J., CLARK S. DNA methylation changes in leukemia. Cancer Biology. 12: p. 347–357. 2002. [ Links ]
7. YANG Y., TAKEUCHI S., HOFMAN W., IKEZOE T., VAN DONGEN J., SZSZEPAMS KI T., BARTRAM C., YOSHINO N., TAGUCHI H., KOEFFLER P. Aberrant Methylation in promoter–assocaited CpG islands of multiples genes in acute lymphoblastic leukemia. Leuk Res. 30(1): p. 98–102. 2006. [ Links ]
8. TAKAHASHI T., SHIVAPURKA N., RIQUELME E., SHIGEMATSU H., REDDY J., ZHOU X., BEKELE N., GAZDAR A., WISTUBA I. Aberrat promoter Hypermethylation of múltiple genes in Gallbladder Carcinoma and Chronic Cholecystitis. Clinical Cancer Res. 10: p. 6126–6133. 2004. [ Links ]
9. ESTELLER M., CORN P., BAYLIN S., HERMAN J. A gene hypermethylation profile of human Cancer. Cancer Res. 61: p. 3225–3229. 2001. [ Links ]
10. NEPHEW K., HUANG T. Epigenetic gene silencing in cancer initiation and progression. Cancer Letters, 190: p. 125– 133. 2003. [ Links ]
11. MILLER S.A., DYKES D.D. A simple salting out procedure for extracting DNA from human nucleated cells. Nuc. Ac. Res. 16: p. 1215. 1988. [ Links ]
Recibido: 19/10/07.
Aprobado: 04/08/08.

