











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
PermalinkINTRODUCCIÓN
La arveja (Pisum sativum L.) es una leguminosa autógama de estación fría y anual que se origina en áreas del Medio Oriente, el este del Cáucaso, Irán y Afganistán, y el oeste de la cuenca mediterránea (Smýkal et al., 2011). Su genoma está organizado en siete pares de cromosomas (2n=2x=14) y el tamaño del genoma haploide se estima en 4,45 Gb (Smýkal et al., 2012). Fueron y son hasta el día de hoy una fuente importante de alimento para animales y humanos. La especie es rica en proteínas, almidón de digestión lenta, azúcares solubles, fibra, minerales y vitaminas (Dahl et al., 2012).
Teniendo en cuenta que el aumento de la población mundial requerirá una mayor producción de cultivos es necesario entonces, incrementar la tasa actual de aumento de los rendimientos para satisfacer esta demanda (Tester y Langridge, 2010). Para ello pueden recurrirse a diferentes estrategias.
Utilización de marcadores moleculares
En este contexto, el desarrollo de mapas de ligamiento se constituye en una herramienta útil para maximizar la probabilidad de éxito, ya que son herramientas poderosas para la investigación genética y el mejoramiento, y son el primer paso en: a) el análisis de rasgos cualitativos y cuantitativos; b) la introgresión de genes deseables y loci de rasgos cuantitativos (QTL); y c) clonación posicional o basada en mapas de genes responsables de rasgos económicamente importantes (Semagn et al., 2006). Diferentes tipos de marcadores se han utilizado para desarrollar mapas de ligamiento de densidad moderada en arveja. Loridon et al. (2005), Sun et al. (2014) y Yang et al. (2015) utilizaron SSR (Simple Sequence Repeats); Deulvot et al. (2010) utilizaron SNP (Single Nucleotide Polymorphisms); Mishra et al. (2009) emplearon ISSR (Inter Simple Sequence Repeats) y Barilli et al. (2010) construyeron sus mapas usando STS (Sequence Tagged Sites). Nosotros propusimos el uso de Sequence Related Amplified Polymorphism (SRAP) (Li y Quiros, 2001) para generar una serie de marcadores distribuidos en todos los cromosomas de arveja. Desde su desarrollo, estos marcadores se han empleado en una amplia gama de especies para la estimación de la diversidad genética (Bermejo et al., 2014; Zheng et al., 2017; Kumar y Agrawal, 2017), en la construcción de mapas (Martin et al., 2013; Padmakar et al., 2015) y en análisis QTL (Liu et al., 2016; Martin et al., 2016).
Para su desarrollo se generó una población de mapeo F2 derivada de un cruzamiento inicial entre dos cultivares DDR11 (The Indian Council of Agriculture Research) y Zav25 (material local derivado de nuestro programa) que son contrastantes para la mayoría de las características relacionadas con el rendimiento, como el número de vainas y semillas por parcela. Se evaluaron un total de 25 combinaciones de cebadores SRAP en plantas F2 y en ambas líneas parentales, generando 208 bandas/marcadores polimórficos. Este primer mapa de ligamiento (Guindón et al., 2016) constó de 112 marcadores genéticos distribuidos en 7 grupos de ligamiento (LG), que cubren un total de 528,8 cM. La longitud de los LG varió de 47,6 a 144,3 cM (media 75,54 cM), con 9 a 34 marcadores.
Posteriormente y en un trabajo en conjunto con el Dr. Thomas Warkentin del Crop Development Centre, Department of Plant Sciences, de la Universidad de Saskatchewan, (Canadá), se profundizó en el desarrollo de un mapa de ligamiento genético usando SRAP, SSR y SNP para identificar QTL que controlan los caracteres relacionados con el rendimiento. Una población F2 y sus familias F2:3 derivadas de un cruce inicial entre cvs. Explorer (desarrollada por Svalof Weibull en Suecia), variedad a semi-áfila, con color verde de cotiledón y vainas y granos de tamaño intermedio y DDR14 (desarrollada por Indian Council of Agriculture Research), foliosa, con cotiledón color amarillo, vainas y granos de gran tamaño, fueron evaluadas con SRAP, SSR y técnicas GBS, que demostraron ser eficientes, generando un conjunto de 872 marcadores polimórficos para mapeo de ligamiento. El mapa resultante constó de 128 genes marcadores distribuidos en 9 grupos de ligamiento (LG) (Figura 1 a y b), que cubrieron 655,5 cM. La longitud de los LG osciló entre 49,1 y 114,8 cM, con 8 a 26 marcadores. La detección de QTL fue realizado utilizando el método mapeo de intervalo compuesto (CIM). Se detectaron un total de 45 QTL a través del generaciones y ambientes evaluados. Todos ellos fueron QTL importantes que explicaron más del 10% de la variación fenotípica (Guindón et al., 2019b).

Figura 1 Mapa de ligamiento, con nombres de los marcadores a la derecha de cada LG (grupo de ligamiento) y distancias del mapa (en cM) a la izquierda. Marcadores con niveles significativos de distorsión por segregación se indican con asteriscos. (Figura tomada de Guindón et al. 2016 CBAB)
Estos estudios nos permiten aplicar estos conocimientos en el programa de mejora asistida por marcadores moleculares.
Selección eficiente de progenitores a hibridar: Uso de BLUP
Diferentes criterios han sido empleados para la selección de variedades a hibridar para generar una población F2 susceptible de ser seleccionada. Un primer criterio está basado en la explotación de la heterosis para diferentes caracteres agronómicos luego de la hibridación ya que existe una correlación positiva entre heterosis y alta frecuencia de líneas recombinantes transgresivas (Rieseberg et al., 1999; Espósito et al., 2014; Joshi et al., 2015, Guindón et al., 2018). Un segundo criterio consiste en la selección de variedades basado en sus valores fenotípicos, pero no es el más adecuado al estar estos valores fenotípicos influenciados por el efecto de las desviaciones ambientales. Un tercer criterio está basado en la utilización de valores genotípicos. Lorenzana y Bernardo (2009) evaluaron la precisión de diferentes métodos para la predicción de valores genotípicos y concluyeron que el enfoque BLUP (Best Linear Unbiased Prediction) es el mejor método para predecir los valores genotípicos en poblaciones biparentales ya que se corrigen mejor las variaciones extrañas, se pueden analizar datos desbalanceados y se puede incorporar la información del pedigrí (Piepho et al., 2008). Guindón et al. (2018) aplicaron BLUP para la predicción de valores genotípicos utilizando datos morfológicos de diferentes años correspondientes a las variedades progenitoras, la generación F1 y las poblaciones F3 ya que la población F2 al no poderse replicar fue imposible establecer el modelo lineal para la obtención de los valores BLUP. Este análisis permitió la corrección por efectos ambientales. Estas estimaciones se utilizaron para el análisis genético de diferentes caracteres. Se observó heterosis para número de vainas (27,1%) y número de semillas (23,3%), caracteres que tienen un efecto directo sobre el rendimiento. Un análisis de componentes principales (PCA) se realizó utilizando datos de BLUP para obtener una representación gráfica de la estructura de relaciones de las 110 familias F3 (Figura 2). Se calcularon las distancias de Manhattan y la matriz de distancias se sometió a un análisis de agrupamiento utilizando el promedio ponderado.
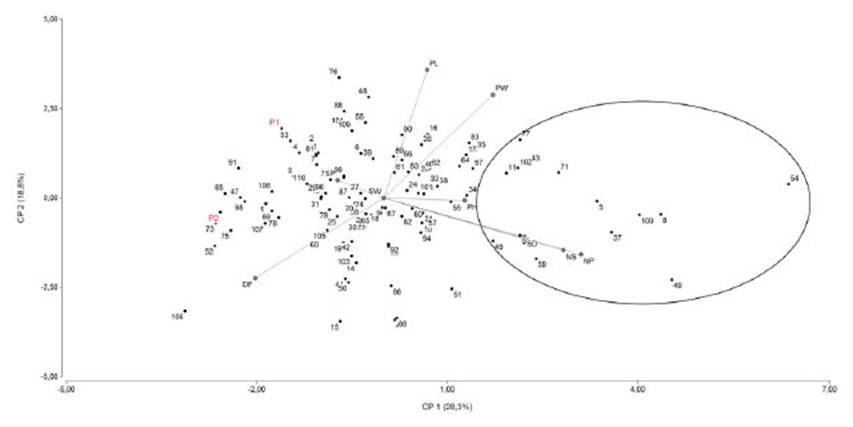
Figura 2 Relaciones entre las 110 familias F3 derivadas de Explorer (P2) y DDR14 (P1) en base al análisis de componentes principales. Las familias dentro del círculo son transgresivas para número de semillas y vainas. La posición de los padres es indicada en rojo.
Mediante el uso de BLUP y de un análisis de componente principales, fue posible elegir familias con buen desempeño lo que muestra la importancia de estas metodologías para los programas de mejora de arveja.
Uso del cultivo in vitro
En los programas de mejoramiento se requiere una gran cantidad de individuos F2 para realizar el proceso de selección correctamente, pero a menudo hay pocas plantas disponibles ya que ciertos cruzamientos producen pocas semillas F1, probablemente debido a diferencias en la estructura de la flor que dificultan la castración manual. Para obtener más semillas F2, es necesario entonces proceder a incrementar artificialmente el número de plantas F1. Se han reportado estudios de regeneración in vitro en arveja, utilizando diferentes explantos, como nudos cotiledonales (Rajput y Singh, 2010), cotiledones (Pniewsky et al., 2003), folíolos inmaduros (Fujioka et al., 2000), embriones cigóticos (Sánchez y Mosquera, 2006) y semillas maduras (Zhihui et al., 2009). Espósito et al. (2012) desarrollaron un protocolo rápido, eficiente y reproducible para la regeneración de brotes in vitro y el enraizamiento de dichos vástagos para la obtención de múltiples genotipos F1 usando semillas maduras (Figura 3).

Figura 3 Múltiples vástagos desarrollados de la semilla original. de la enfermedad (Porob et al., 2017).
Las plántulas regeneradas se trasplantaron al suelo en macetas, encontrándose que el 60% de los vástagos regenerados enraizaron en un período de seis semanas. Generalmente, las tasas de mortalidad se debieron a tallos y raíces débiles que no pudieron adquirir los nutrientes del suelo cuando son transferidas del medio agarizado. Las plántulas que desarrollaron normalmente se trasplantaron a macetas y en invernadero cultivándose hasta la madurez. Este procedimiento se puede utilizar en programas de mejoramiento lo que permitirá trabajar con más plantas siempre que los cruces tengan poca producción de semillas.
Uso de fenotipificación digital
La fenotipificación de plantas vincula la genómica con la ecofisiología y la agronomía. Por lo general, se realiza mediante tecnología no destructiva, automatizada y basada en imágenes, y genera información para la eficiente caracterización digital y que se puede realizar durante la regeneración periódica y rutinaria de accesiones en colecciones de germoplasma. Gatti et al. (2017) estudiaron 92 accesiones del género Pisum de diferentes especies y subespecies durante dos ciclos de cultivo midiendo caracteres asociados al tamaño y color de semillas y vainas utilizando imágenes digitales. Estas características obtenidas por fenotipificación digital, demostraron ser marcadores adecuados para la evaluación de la diversidad genética y útiles en el análisis evolutivo, permitiendo la discriminación de las principales especies silvestres y cultivadas del género Pisum. También se la ha utilizado para fenotipificar varios parámetros del estado fitosanitario, como contenido de clorofila (Dutta Gupta et al., 2013), contenido de nitrógeno (Vollmann et al., 2011) y estado de la enfermedad (Porob et al., 2017).
Durante el almacenamiento de la arveja pueden ocurrir pérdidas significativas en el color, debido a la pérdida de clorofila o al blanqueado de las semillas y este deterioro influye en la comercialización y en la decisión de compra por parte de los consumidores. La magnitud de esta pérdida de color puede ser estudiada a partir de la técnica de envejecimiento acelerado, que implica someter a las semillas a condiciones severas de temperatura y humedad relativa y usar el fenotipificación digital es para medir dicho deterioro. Guindón et al. (2019 a), demostraron la existencia de un comportamiento diferencial de las variedades en el mantenimiento del color verde de las semillas mediante esta metodología, pudiendo así también ser aplicada en procesos de selección durante la obtención de nuevas variedades.

