











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La microangiopatía trombótica (MAT) es una condición clínica rara que se caracteriza por anemia hemolítica, plaquetopenia y lesión de órgano,1 incluida la injuria renal aguda (IRA) que se da por la extensa red capilar que predispone al daño endotelial y oclusión vascular.2
El SUH se clasifica, según el Grupo Internacional de SUH, en típico, atípico o asociado a enfermedades, infecciones, déficit de cobalamina, fármacos, embarazo, mutación del gen DGKE, formas familiares de herencia autosómica dominante o recesiva y aquellas de causas no identificadas.3El SUHa se considera una enfermedad ultrarara causada por alteraciones genéticas en la vía del complemento (mutaciones en factor H, factor I, factor B, MCP, trombomodulina (THBD), anticuerpos del factor H y proteínas relacionadas al factor H (CFH), que se expresan en las superficies celulares provocando lesiones microangiopáticas sistémicas.4 La prevalencia es de 3.3 pacientes por millón de habitantes, con un comportamiento progresivo y fatal.4-5) El SUHa autosómico recesivo ocurre en la infancia, el pronóstico es malo y la tasa de mortalidad es entreel 60 y el 70%, mientras que el autosómico dominante a menudo se presenta en adultos, tiene mal pronóstico y un riesgo de muerte o ERC del 50 al 90%.6- 7
El análisis de pacientes con SUH, en diversos estudios internacionales, ha establecido que aproximadamente el 70% tienen anomalías genéticas identificables que alteran la regulación de la vía alternativa.8 Las mutaciones de estos genes son heterocigotas en un 90% de los casos y la mayoría conduce a la pérdida de la función de la proteína, a excepción de las de C3 y CFB, identificadas en el 15% de los casos de SUH, que son mutaciones de ganancia de función.9-10 En algunos casos, se han identificado autoanticuerpos contra CFH adquiridos, con más del 90% de estos pacientes homocigotos para un polimorfismo de los genes CFH R3 o CFH R1.11-13
La presentación clínicaes variable, puede estar seguida de eventos tales como infecciones, vacunaciones o embarazos, considerados como eventos gatilladores.12 La edad de presentación varía, sin predominio en el sexo. La clásica triada de trombocitopenia, anemia hemolítica mecánica, con presencia de esquistocitos e IRA,son los elementos característicos de esta enfermedad.(3) Aunque las lesiones de SUHa afectan, predominantemente, a los vasos renales, la naturaleza difusa del fenómeno MAT conduce a la participación de la microvasculatura de otros órganos, el 63% de los casos tiene compromiso extrarrenal,1) siendo más frecuentes los neurológicos. El infarto de miocardio se ha descrito en hasta el 3% de los pacientes con SUH y se puede relacionar con muerte súbita.14-15
CASO CLÍNICO
Femenina, 19 años de edad, con antecedentes de síndrome Alagille y ERC por agenesia renal derecha. Se evalúa para trasplante renal (TxR) preemptive (clearance de creatinina 22,1 ml/min) con donante vivo. Donante: femenina (madre), 41 años de edad sin antecedentes patológicos, crossmatch contra donante por citotoxicidad negativa, 2 missmatch, sin contraindicación. Previo al TxR presentó dermatosis perforante urémica por lo que inició diálisis.En cirugía se realizó lavado del riñón con HTK, inducción con THBD y corticoides, tiempo de isquemia fría de 30 minutos y tiempo de anastomosis de 30 minutos, cirugía sin complicaciones.Evoluciona posoperatorio con anemia aguda (hematocrito del 20%, con previo de 26%), ecografía normal, requiriendo transfusión de hemoderivados e inicio de tacrolimus (FK), micofenolato sódico y corticoides. Al quinto día, con retraso funcional del injerto (RFI), Cr. 4,7 mg/dl, FK 2 ng/ml, doppler renal con índice de resistencia elevado 0,8, radiorrenograma con fase parenquimatosa y de eliminación plana, compatible con severo compromiso del injerto renal. Se decidió biopsia renal que informó necrosis tubular aguda (NTA) y ausencia de rechazo (Figura 1). Se aumentó dosis de tacrolimus (objetivo de dosaje 8 a 10) y completó dosis de timoglogulina total de 5,61 mg/kg.

Persistió con RFI, nivel de FK 6,9 ng/ml, anticuerpos por Luminex negativo y ecografía renal sin cambios. Se realiza segunda biopsia renal que informó mesangiolisis, aneurismas de capilares glomerulares con C4d negativo, inmunofluorescencia (+) para fibrina y sin rechazo, hallazgos compatibles con MAT (Figura1). Parámetros de hemólisis: haptoglobina 21mg/dl, LDH 426U/L,pese a ausencia de esquistocitos en frotis de sangre. Ante los hallazgos clínicos y en biopsia, se sospecha MAT, probablemente secundaria a tacrolimus,por lo que se suspende y se rota a belatacept e inició de plasmaféresis con previa toma de muestra para ADAMTS 13, con ulterior mejoría de hemólisis.Intercurrió con neumonía lobar, la cual se trató, seguido de compromiso severo del hepatograma (TGO 119 UI/dl, TGP 136 UI/dl, FAL 1072 U/l, BT 62,9mg/dl, BD 48,7 mg/dl, BI 14,7 mg/dl), sin posibilidad de biopsia hepática transyugular por situación clínica.
Dada la persistencia de RFI(día +30), asociado a proteinuria de novo (5,98 gr), se realizó tercera biopsia renal que informó NTA, descartando recidiva de enfermedad glomerular. Evolucionó con crisis convulsivas, se interpretó como asociada a hiponatremia, posteriormente presentó sangrado en sitio de punción de biopsia renal previa, que evidenció en la cirugía sangrado en napa. En el posoperatorio inmediato presentó plejía braquiocrural, tomografía normal y recuperación a las 24 horas del foco motor.
Dados los eventos neurológicos (convulsión, plejía), disfunción renal, anemia con requerimiento transfusional, hemorragias espontáneas sin sitio de sangrado evidente y sin alteración de la coagulación (descartándose PTT: ADAMTS 13 en 13,35%), hemorragia alveolar y todo asociado a descenso de haptoglobina, presencia de esquistocitos aislados en nuevo frotis y hallazgo de MAT en biopsia renal, se solicitó estudio genético para SUHa por fuerte sospecha y se administró Eculizumab 900 mg. Sin embargo, evoluciona tórpidamente con falla multiorgánica y óbito, con necropsia posteriormente.(Figura 2)
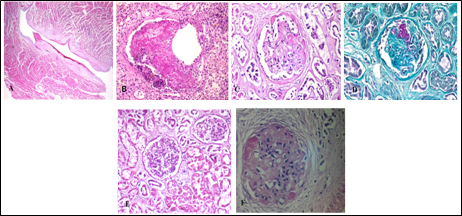
Figura2. Informe de necropsia - (a)Tromboembolia de reciente data a nivel de ventrículo derecho, signos multifocales de isquemia miocárdica; (b) pulmón izquierdo con microtrombos de fibrina en vasos de pequeño calibre; (c) y (d) riñón nativo: glomeruloesclerosis global difusa, microtrombos de fibrina en capilares glomerulares microtrombos (positivos para fibrina con tinción de Fraser-Lendrum), escleroatrofia moderada a intensa; (e) y (f) riñón trasplantado: glomérulos congestivos y permeables, necrosis tubular intensa, cilindros hialinos y pigmentados. Sin signos de rechazo agudo. C4d fue negativa en capilares peritubulares y glomerulares, presencia de material fibrino hemático en organización (hematoma) perirrenal
Estudio genético de paciente,THBD: variante heterocigota c.1357G >A p.(Gly453Arg) y deleción heterocigota de CFHR3 y CFHR1. Clasificándose como variante de significado incierto (clase 3) según Centogene y ACMG. Se completó estudio genético de los padres con previo consentimiento informado: padre: mutación en THBD (variantesignificado incierto), madre: variante del gen CFHde significado incierto, C5b-9 concentración: 496,6 ng/ml.
DISCUSIÓN
En estudios recientes evidenciaron la recurrencia postrasplante del SUHa en más del 70% de los receptores, con diferentes porcentajes de presentación en los genes del complemento. La MAT,generalmente, se desarrolla en el período posterior al trasplante temprano, pero también puede aparecer entre los dos y seis años posteriores al trasplante, por lo que no son claros los factores de riesgo involucrados para su presentación(Tabla 1).16) La lesión por isquemia-reperfusión, el rechazo agudo, las infecciones virales y los fármacos inmunosupresores, como los inhibidores de la calcineurina o sirolimus, se han relacionado con el desarrollo de MAT después del trasplante renal.
Según Loirat et al., en un estudio de cribado en 273 pacientes diagnosticados de SHUa de forma esporádica y familiar, la mayoría de los pacientes presentaban mutaciones o polimorfismos de los genes de factor H, lo que sugiere una predisposición genética. Los pacientes con mutaciones de CFH o THBD tuvieron el inicio más temprano y la mortalidad más alta. Las mutaciones de la proteína cofactor de membrana (MCP) se asociaron con el mejor pronóstico. La remisión inducida por terapia con recambio de plasma se observó entre el 55 al 80% de los episodios en pacientes con mutaciones o autoanticuerpos CFH, C3 o THBD, mientras que los pacientes con mutaciones de CFI no respondieron de la misma manera.17
En un estudio realizado por Delvaeye et al., donde secuenciaron el gen de THBD en 152 pacientes con SUHa y en 380 pacientes controles, los resultados obtenidos fueron que 7 pacientes no relacionados tenían seis mutaciones heterocigotas y sustitutivas del gen, concluyendo que existía alrededor del 5% de mutaciones del gen de la THBD asociado a SUHa.18
Una reciente publicación por el grupo francés de Fakhouri et al., describió los hallazgos genéticos en una cohorte de pacientes definidos como SUHa secundario. En ella demuestran que las mutaciones raras, de significado incierto o consideradas como no patógenas, y los polimorfismos, eran frecuentes en estos pacientes con SUHa. No obstante, esas mismas mutaciones fueron halladas en la población sana testigo, y las mutaciones del factor H, factor B y factor I eran las halladas en los SUHa clásicos (no secundarios). Fueron tratados con Eculizumab los casos más severos y los que requirieron diálisis.19
En nuestro caso se encontró una mutación del gen de THBD heredada de su padre, es una variante de significado incierto pero asociada a mayor susceptibilidad para el desarrollo de SUHa, por lo tanto es autosómica dominante. Por otra parte, la madre presentaba una variante en heterocigosis en el gen CFH de significado incierto reportada previamente en la literatura en pacientes con SUHa.Dado que ambas mutaciones son halladas en la paciente en heterocigosis, no pueden definirse ciertamente como patógenas, no obstante, la manifestación clínica y los hallazgos en la necropsia de trombosis sistémica hacen suponer la predisposición tanto en receptora como en el órgano donado a ser pasible de injuria endotelial por activación de vía de complemento, en contexto apropiado y en las circunstancias desencadenantes, tales como la injuria de reperfusión, el trasplante en sí mismo, los mecanismos de inmunidad innata, el uso de tacrolimus y las infecciones.
CONCLUSIONES
Se requieren más estudios clínicos y genéticos en conjunto para definir el rol patógeno de mutaciones no clásicas o en heterocigosis, camino que creemos es necesario recorrer para dar explicación a estos procesos de tal gravedad y por la rapidez en la forma de instalarse, así como en la refractariedad al tratamiento que requieren diagnóstico y tratamiento precoz para evitar casos fatales.
Conflicto de intereses: Los autores declaran no poseer ningún interés comercial o asociativo que presente un conflicto de intereses con el trabajo presentado.

