











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Animals are affected by many pathogens, causing economic losses due to direct costs for decreased performance and production and additional costs for veterinary assistance and extra care of ill animals. Importantly, some of them are zoonotic and yet receive little attention. This is the case for bovine leptospirosis, a widespread and potentially fatal zoonosis disease. It is caused by bacteria of the genus Leptospira spp. and produces abortions, the birth of weak calves, infertility, mastitis, and agalactia. Moreover, chronically infected animals with no symptoms act as reservoirs spreading pathogenic leptospires through urine, contaminating water sources, and soil (Levett 2001).
Caprine brucellosis is an endemic zoonotic disease with a wide distribution around the world. In Argentina, the presence of the disease is strongly distributed in areas of high goat production, such as in goat breeding regions for subsistence self-consumption, negatively affecting public health (Rossetti 2017).
Leishmaniasis is another group of zoonotic diseases caused by flagellated hemoparasites of the genus Leishmania spp., which are transmitted through the bite of female phlebotomine sandflies to humans and other mammals. The disease is endemic in 98 countries and affects 12 million people around the world. According to the World Health Organization, there are 350 million people at risk, and 2 million new cases are reported each year (World Health Organization 2010). Domestic dogs are very susceptible to this parasite and play an important role in the transmission to humans since they act as reservoirs of infection in both rural and urban areas (Dantas-Torres 2007).
Among diseases of economic importance, it can be mentioned the equine paratyphoid and avian coccidiosis. The former is an infectious disease caused by Salmonella enterica subsp.enterica ser Abortusequi (S. Abortusequi), that manifests mainly as abortion in the mare, orchitis in stallions, and septicemia and polyarthritis in foals. It is a re-emerging disease, affecting negatively the equine production in Argentina (Bustos et al. 2020).
Avian coccidiosis is caused by a protozoan parasite of the genus Eimeria spp. and is a highly contagious intestinal parasitosis that involves important losses in the production of avian meat. The economic burden is estimated at more than USD 3 billion in the USA and in Europe more than 2 billion pounds (£) per year (Dalloul and Lillehoj 2006; Pastor-Fernández et al. 2018). Seven species of Eimeria affect chickens, each demonstrating a high degree of site-specificity in the chicken gut and different pathogenicity.
These five pathogens of veterinary significance are diagnosed conventionally, which is mainly based on their isolation in culture (Leptospira spp. and Brucella melitensis) (Godfroid et al. 2011) or direct observation under microscopy (Eimeria spp.) and through serological diagnosis (Leptospira spp., Leishmania spp.). Conventional techniques include the Microscopic Agglutination Test (MAT), bacteriological isolation from urine during leptospirosis acute infection or from tissues during brucellosis infection, and the direct observation of Eimeria sp. oocysts in fecal samples, and from the serial scraping method of the intestinal mucosa.
Although these conventional methods have contributed significantly to the control of different diseases, they have some disadvantages such as low sensitivity, being laborious or time-consuming, presenting the risk of bacteriological contamination or the difficulties in the identification of the isolated species. The ability to achieve isolations and characterization of B. melitensis is restricted to laboratories with a high degree of biosafety facilities, given the risk of working with highly pathogenic agents (Jacques et al. 2010). Furthermore, in the case of Eimeria spp. identification of species by conventional methods such as oocyst morphology and pre-patent period can be difficult, even for an expert (Kumar et al. 2013).
Molecular diagnosis in the last decades has provided enormous advances. In this sense, downstream application such as PCR, and its variants, i.e. multiplex- PCR, nested- PCR, LAMP, and qPCR among others usually are highly sensitive and specific, allowing the detection of low amounts of microorganisms, and also help in typing or phylogenetic studies. Some of them are ongoing works for some pathogens. This is the case for LAMP for the detection of Leptospira spp., multiplex-PCR for Eimeria spp. (unpublished data), and PCR for Salmonella spp. from the vagina, placenta, and different organs of equine aborted fetuses as a fast approach for equine paratyphoid diagnosis (Bustos et al. 2020).
Beyond the molecular method, the quality and quantity of nucleic acid extraction from samples are a key step. The success of PCR-based technologies depends on the process of extraction and purification of DNA that eliminates contamination and inhibitors, keeping good quality and integrity of the target sequence to be amplified. Although the first nucleic acid isolation was achieved 150 years ago, there is no general Gold- Standard for downstream applications. Chelex-100 resin has been used since the '90s for diagnostic (Noh et al. 2019; Chen et al. 1991) and other purposes (Gill et al. 1992) given that it is a rapid, easily performed, and non-expensive method from a variety of biological samples such as semen, blood, tissues, and milk among others, with different sensitivities depending on the pathogen (Natarajan et al. 2018).
Since molecular diagnosis for the five pathogens mentioned above would complement conventional diagnosis, allowing typing in some cases, testing the best DNA isolation method is crucial, and would contribute to the improvement in control and prevention strategies. The goal of the present work was to compare the in house 5% Chelex-100 chelating resin (M1) with three methods such as Whatman® cards (M2), phenol-chloroform organic solvent (M3), or commercial kits (M4) to determine which one yields effective results for the molecular diagnosis for three bacteria: Leptospira interrogans serovar Pomona Pomona, B. melitensis, and S. Abortusequi, and two parasites of the genus Eimeria spp. and Leishmania spp. from distinct types of samples, i.e. urine, organs, semen, blood, and intestinal mucosa, respectively.
Materials y methods
Samples for DNA isolation
Animal samples were experimentally infected with the following pathogens: the bacteriaLeptospira interrogans serovar Pomona Pomona and S. Abortusequi, and the parasite Eimeria spp. For the former, bovine urine was artificially contaminated with Leptospira interrogans serovar Pomona Pomona culture in EMJH medium, as previously reported by Hamer et al. 2019. This had 2 x 10. leptospires per mL according to McFarland Scale. Tenfold serial dilutions of L. interrogans serovar Pomona Pomona were prepared from 10-1 (2 x 10. leptospires/ mL) to 10-10 (2 x 10-2 leptospires/ mL) as follows: firstly, 4.5 mL of fresh urine was contaminated with 0.5 mL culture. Then 100 µL of each dilution was mixed with 900 µL of urine and was subjected to DNA isolation.
Semen pool sample from equines was artificially contaminated with different concentrations of the strain S. Abortusequi UBA1174/15. Briefly, the bacterium was incubated into soy trypticase agar at 37°C ON, and then, a single colony was resuspended in PBS buffer. Bacteria suspension turbidity was adjusted at 0.5 McFarland Standard (approximately 1.5 x 10. cells/ mL) and the surface viable count (Miles, Misra, and Irwin 1938) was used to determine the number of colony-forming units (CFU/mL) in triplicate. The suspensions were serially diluted 1:10 starting from 1:100 to 1:1,000,000 and 100 µL of each dilution was added to 900 µL of an equine semen pool sample (final volume 1,000 µL). Two hundred µL of each contaminated sample were subjected to DNA extraction.
Oocysts of Eimeria tenella were isolated from the caeca of experimentally infected birds as previously described (Chapman and Shirley 2003). Afterwards, the sporulation of isolated oocysts was carried out in potassium dichromate at 2% (Sigma, USA) for 3 days at 28 °C. They were bleached with sodium hypochlorite and washed 3 times with TE buffer (10 mM Tris/ HCl, Promega, USA), 1 mM EDTA (Promega, USA) in deionized water (pH 8.0) using a bench centrifuge at 500 . for 8 min. Finally, oocysts were pelleted, the supernatant was discarded and resuspended in equal volume of Buffer TE and covered with equal volume of 425- 600 µm glass beads (Sigma, USA), previously sterilized by autoclaving at 121 °C, 15 min. To break the oocysts, up to 5 of 3 min at a maximum speed were performed with vortex mixer Fbr® (Decolab). The cut-off was determined by calculating the percentage of rupture by counting the entire oocysts under a microscope in the Neubauer chamber and more than 85 % of rupture was achieved. Supernatant from the disrupted oocysts was centrifuged at 14,000 . for 10 min and was subjected to DNA isolation from a total of 24,000 caecal oocysts.
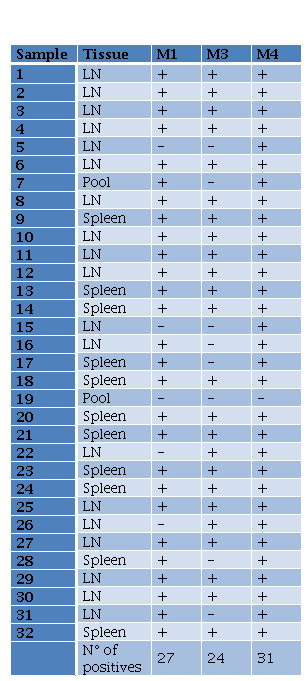
DNA from Brucella melitensis and Leishmania infantum were isolated from naturally infected animals. B. melitensis was isolated from 32 serologically positive goats. Samples from spleen and lymph nodes were obtained, but lymph nodes were exclusively used (19 of 32 total samples) and were subjected to DNA extraction. For the evaluation of the performance, a proportions test was applied to the different methods.
A blood sample from a L. infantum positive canine was collected through venipuncture and stored at −20 °C in tubes with sodium citrate 3.8 % until use. The diagnosis was made using the KalazarDetect™ Canine Rapid Test serological kit (INBIOS, USA). 100 µl of blood was subjected to DNA isolation.
DNA extraction methods
Four methods were employed depending on the pathogen and the type of sample (Table 1).
Table 1. DNA extraction methods performed for different pathogens
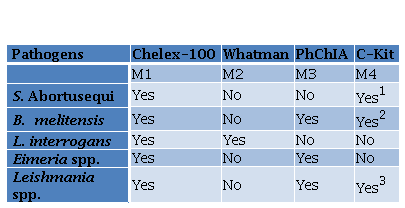
1PetNAD Nucleic Acid Co-prep kit
2QIAamp® DNA Mini Kit (QIAGEN)
3DNeasy® Blood & Tissue (QIAGEN®).
Method 1 (M1): the Chelex-100 resin (BioRad) was prepared and employed according to manufacturer instructions and adapted to each type of sample (Table 2). Briefly, the different samples were incubated with distinct volumes of resin Chelex-100, as described in Table 2, followed by two incubation temperatures of 56 °C and 100 °C, except for equine semen and canine blood samples. Afterwards, a vortex step for urine and caecal mucosa samples followed the incubation step. Finally, a centrifugation step was performed to recover the supernatant, containing DNA, applying different conditions depending on the type of sample. Method 2 (M2): FTA Elute modified protocol using FTA Whatman Classic Card (GE Healthcare Life Sciences) was performed. A punch of FTA card was taken using 3 mm UniCore and was placed in a 1.5 µL microcentrifuge tube. 50 µL of urine sample was transferred and incubated for 5 min and the liquid was discarded. Then, it was dried at 100 ºC for 5 min and 500 µL of DNase free water was added and vortexed for 5 s. After the supernatant was discarded, 30 µL of water was added. After centrifugation at 9,000 g for for 10 s, the samples were heated at 100 ºC for 15 min and vortexed for 30 s. Finally, samples were centrifuged for 30 s and the supernatant was preserved at -20 ºC until use. Method 3 (M3): The organic solvent phenol: chloroform: isoamyl alcohol (PhChIA), 25: 24: 1 (Thermo Fisher Scientific, USA) was used for E. tenella, L. infantum, and B. melitensis. For the latter, 50 mg of tissue was placed in a 1.5 μl microtube with 750 μl of lysis buffer (20 mM Tris-HCl (Promega, USA), 5 mM EDTA pH 8 (Promega, USA), 400 mM NaCl, 1% SDS (Promega, USA), proteinase k 400 μg/ ml (Promega, USA) and incubation was carried out for 8 h at 55 °C (Vejarano et al. 2013). E. tenella sporulated oocysts were broken with glass beads as mentioned above and following supernatant recovery from the disruption, it was mixed with 0.33 vol of 10% sodium dodecyl sulphate solution (Promega, USA) and incubated at 37 °C for 2 h in the presence of 100 μg /ml of proteinase K (Promega, USA) as described Blake et al. (2003). Thereafter, each sample was centrifuged at 10,000 g for 1 min.
Table 2. Modified Chelex Protocol according to the type of sample.
Table 2. Modified Chelex Protocol according to the type of sample
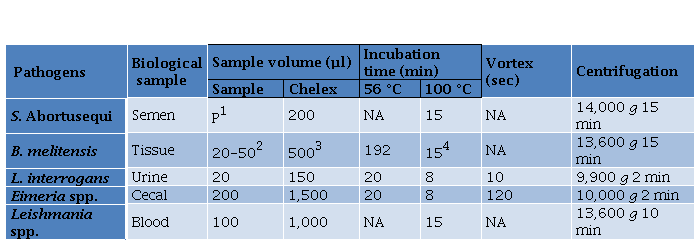
1200 µl of semen sample was centrifuged at 1,500 g for 5 min and the pellet (P) was kept,
2500 µL of DNAse/ RNAse was added to 20 to 50 mg and were homogenized
320 μl of proteinase K (20 mg/ ml) (Promega, USA) was added
4After the incubation, 2.5 volumes of 100% ethanol was added, centrifuged at 13,600 g for 15 min and resuspend in 70% ethanol; NA: not applied.
The lysate solution of sporozoites of Eimeria sp. and 100 µl blood of a L. infantum positive dog DNA from the three pathogens was extracted using the organic solvent as follows: 1 vol of PhChIA was added to the DNA suspended in each supernatant and vortex for 20 s; then, the sample was centrifuged at 16,000 . for 5 min and the supernatant was removed, 0.5 Vols of 7.5 M of sodium acetate (Biodynamics) and 2.5 Vols. of absolute ethanol (Merck). To allow precipitation of the DNA, an overnight incubation was performed at –80 °C. The following day the sample was centrifuged at 16,000 . for 30 min at 4 °C, washed with 70% of ethanol (Merck), and finally the DNA was resuspended in 200 µL of buffer TE (Blake et al. 2003), but not the L. infantum DNA, which was resuspended in 50 µl of H.O milli-Q ®. Method 4 (M4): three different commercialized kits were employed depending on the type of sample. From tissues of serologically positive animals for B. melitensis the QIAamp DNA Mini Kit (QIAGEN ®) was used, after homogenizing 20- 50 mg of tissue as described in the previous in M3. From 200 µl of equine semen the PetNAD™ Nucleic Acid, Co-prep kit (GeneReach, USA) was used for the later detection of S. Abortusequi by PCR. From blood samples from a Leishmania infantum positive dogs, the DNeasy® Blood & Tissue (QIAGEN ®) kit was used. In all cases DNA extraction was performed according to the manufacturer recommendations without modifications.
DNA solution extracted by the four extraction methods was preserved at -20 ºC until use except for Leishmania DNA, for which aliquots were thawed and re-frozen at 30, 60, and 90 days.
Dilutions of the DNA were performed for L. infantum and E. tenella. For the former, ten serial dilutions of the DNA were performed, using a dilution factor of 0.33 (from 1 to 1 x 10-6). For E. tenella, DNA was serially diluted (1: 2), starting with a total of 24,000 or 1,200 oocysts for M3 and M1, respectively. Theoretical oocysts were calculated by dividing the oocysts by the volume of resuspension used, i.e. 200 µl for M3 (1,200 oocysts/ µl) and 350 µl for M1 (34 oocysts/ µl). The initial theoretical number of oocysts per PCR was adjusted starting at 300.
Polymerase Chain Reactions (PCRs)
To detect DNA from Leptospira interrogans serovar Pomona Pomona, S. Abortusequi, B. melitensis, Leishmania spp. and Eimeria spp. PCRs were performed with specific primers previously reported by Bal et al. (1994); Malorny et al. (2003); Bricker and Halling (1994); Rodgers et al. (1990) and Schwarz et al. (2009), respectively. Conditions and previously described primers are listed in Table S1 of the supplementary data.
The volume of DNA used as template was 3 or 4 μL for a final volume of 50 µl except for Eimeria that was ranged between 1.5 and 8 µl of genomic DNA template, depending on the sample. PCR products were resolved by 1, 1.5, or 2 % agarose (Biodynamics) gel with ethidium bromide electrophoresis and visualized under UV light with a transilluminator (Uvi Tec trans-illuminator BTS-20.M). Band sizes were determined with the following molecular weight ladder of 100bp precision (Productos Bio-Lógicos, Buenos Aires, Argentina), GeneRuler 100bp® DNA Ladder (Promega), and GenRuler™ 1 kb (ThermoScientific), as indicated.
Results
For leptospirosis, DNA extraction methods M1 (Chelex-100) and M2 (Whatman cards) were compared for the detection of pathogenic leptospires in contaminated urine. It was observed that M1 had a better performance than M2 since leptospiral DNA was detected up to the sixth dilution (10-6), representing an approximate of 2 x 10. leptospires/ mL (Fig. 1A), whereas the M2 detected up to the dilution 10-3 (2 x 10. leptospires / mL) (Fig. 1B).
In the case of B. melitensis, M1 (Chelex-100), M3 (PhChIA), and M4 (C-kit) methods were compared for molecular detection of positive samples. The proportions test applied did not distinguish a greater capacity for detection by any particular method. Results showed a greater detection trend using the commercial kit. Of a total of 32 positive tissues, M4 detected the presence of B. melitensis in 97% (31/32), whereas the other 2 methods did not exceed 85% (27/32) (Table 3).
Leishmania DNA was extracted by M1 (Chelex-100), M3 (PhChIA), and M4 (C-Kit) (Fig. 2A). To compare the sensitivity of the extraction methods, serial dilutions of the DNA extracted with each of the three methods were used as a template for the PCR, and the maximum dilution was recorded in which a positive amplification was obtained. Our results showed that Leishmania DNA was detected up to a dilution of 3 x 10-4 with the three methods. Fig. 2B shows, as an example, the amplification curve with M1. Additionally, the DNA stability for Leishmania spp. was evaluated. For this purpose, aliquots of DNA extracted by M1 and M4 were stored at -20 ° C and were thawed and re-frozen at 30, 60, and 90 days. PCR results showed that both extraction methods preserved DNA similarly, given that at 90 days samples amplified up to a dilution of 3 x 10-3, i.e. one order of magnitude less than the samples without thawed and re-frozen.
For S. Abortusequi, similar analytical sensitivities were determined for Chelex-100 (M1) and a commercial kit (M4). Approximately, 1.415 x 10. CFU/mL and 1.415 x 10. CFU/mL of S. Abortusequi were the analytical sensitivity for M1 and M3 methods, respectively (data not shown).
Eimeria genus was detected from caecal oocyst from both methods of DNA isolation PhChA (M3) and Chelex-100 (M1) (Fig. 3A). Afterwards, the analytical sensitivity was assessed for both methodologies by a sensitivity curve. It was performed a serial dilution of the DNA and calculated the theoretical oocysts per PCR (tOP), showing amplification up to 18 tOP for M1 and 9 tOP for M3 (Fig. 3B).
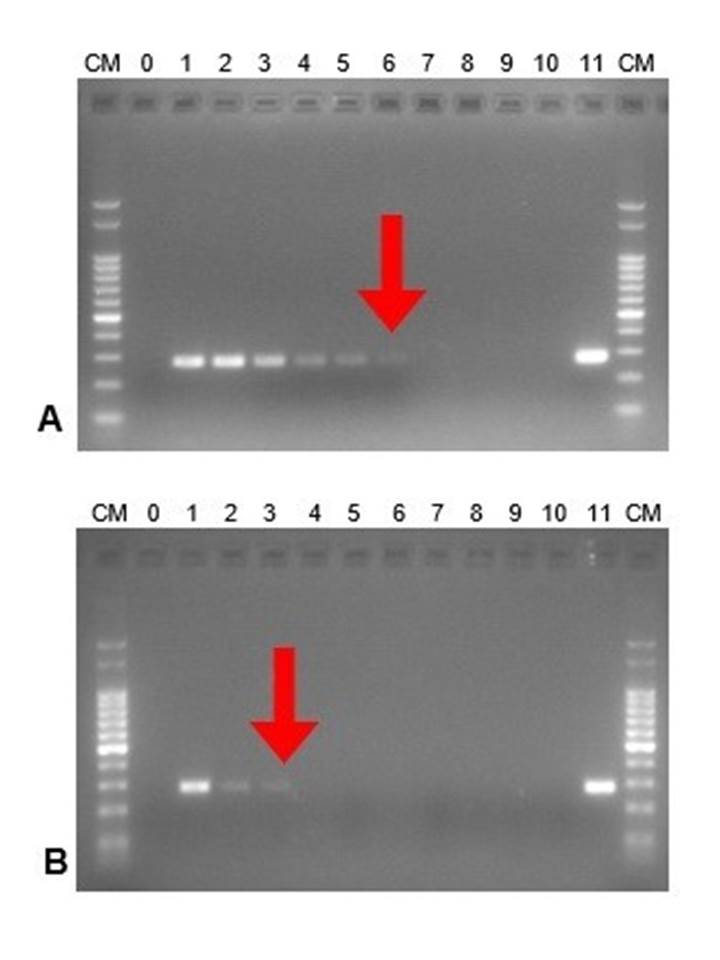
Figure 1. DNA amplification from serial dilutions of pathogenic leptospires (Leptospira interrogans serovar Pomona Pomona). In vitro assay. DNA ladder (CM):100 bp ladder; 0: negative control. Serial dilutions from 1 to 10. 11: positive control. The best dilution obtained is highlighted with a red arrow. A. Extraction method M1 (Chelex-100), B. Extraction method M2 (Whatman cards).
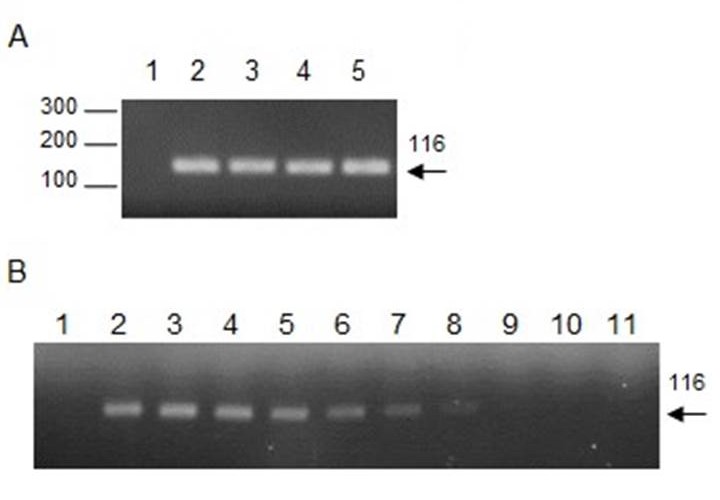
Figure 2. Detection of Leishmania spp. and the sensitivity curve. A. Amplification of DNA fragments of the Leishmania spp. kinetoplast minicircle by PCR using 13A/13B primers. A 116 bp band is observed using DNA extracted with M1 (2), M3 (3), and M4 (4) from the blood of a positive dog of Leishmania infantum (diagnosed by microscopy), and DNA from Leishmania infantum culture was used as the positive control (5) or water as the negative control (1). B. Sensitivity curve. Amplification of serial dilutions 1 (2), 1 x 102 (3), 3 x 102 (4), 1 x 103 (5), 3 x 103 (6), 1 x 104 (7), 3 x 104 (8), 1 x 105 (9), 3 x 105 (10), 1 x 106 (11), using DNA extracted with Chelex-100 or water as negative control (1).
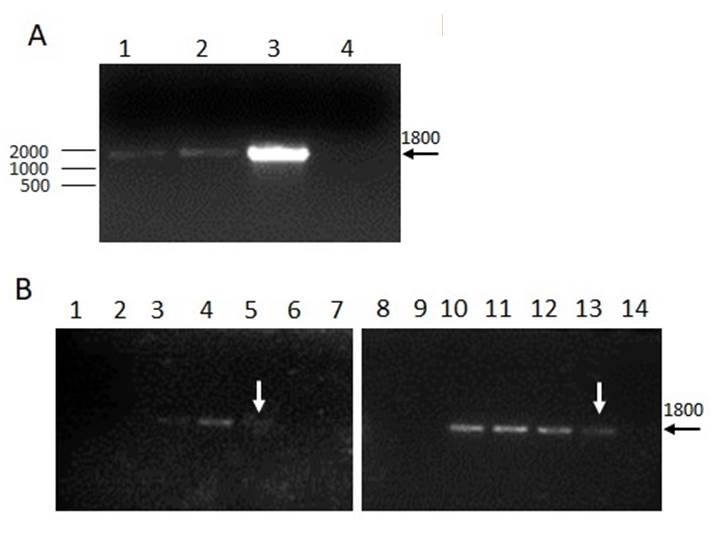
Figure 3. Detection of Eimeria spp. and the sensitivity curve. A. Amplification of an 18S rDNA fragment from Eimeria spp. A clear 1800 bp band is observed using DNA isolation with both methods, Chelex-100 (M1) or PhClAI (M3). DNA from live vaccines was used as positive control (3) or water as the negative control (4). B. Analytical sensitivity. Serial dilutions of DNA (1:2) were performed for isolation with M1 (1 to 7) or M3 (8 to 14). Theoretical oocysts per PCR: 300 (1 and 8); 150 (2 and 9); 75 (3 and 10); 37 (4 and 11); 18 (5 and 12); 9 (6 and 13) and negative control (7 and 14).
Discussion
PCR is a downstream application for DNA detection, which is a versatile designed molecular technique that has enabled the development of diagnostic protocols for different etiological agents. Despite acid nucleic extraction has been achieved many years ago, there is no Gold-Standard method to the present time. Chelex-100 is a styrene-divinylbenzene copolymer containing paired iminodiacetate ions, which selectively chelate transition metal ions. When the resin is mixed with the sample before boiling, the DNA damage is prevented besides DNA polymerase inhibitors are attenuated. This leads to DNA with enough integrity to remain as a template for the amplification of target sequences for diagnosis. The use of the resin is attractive because it is a simple, fast, and economic method especially in low-resource settings (Turan et al. 2015; Ascencio et al. 2019).
The present work shows that DNA extraction with Chelex-100 resin was successfully applied in five animal pathogens where it had not been tested before and can be applied for PCR detection although with slightly different sensitives for each pathogen. Chelex-100 showed better sensitivity than M2 for L. interrogans serovar Pomona Pomona and similar sensitivity for Leishmania spp. when compared with M3 and M4; for S. Abortusequi when compared with M4; and for Eimeria spp. when compared with M3. The performance of DNA extraction based on the use of Chelex-100 for detection and diagnosis of B. melitensis was slightly surpassed by M3 (PhChIA), which is immediately below the cases detected by M4 (commercial kit). The use of the resin is a valid alternative for the detection of Brucella sp. from tissue samples, considering the particularity of being a facultative intracellular bacterium.
Tabla 3. Results for B melitensis PCR from DNA extracted using different methods LN lymph nodes M1M3M4 methods 1 3 and 4
Results for Leishmania spp. are according to Lauchaud et al. (2002), who established a sensitivity of 1 x 10-3 parasites per milliliter for this PCR. Additionally, a recent study performed by Ascencio et al. (2019) has shown that Chelex- 100 resin was applied to extract DNA from whole blood, being useful for the diagnosis of canine leishmaniasis.
For the case of Salmonella, the sensitivity of the analytic detection with M1 was 1.415 x 104 CFU/ mL, slightly lower than M4. Pathmanathan et al. (2003) determined similar sensitivity for final point PCR identifying Salmonella ser. Typhimurium (3 x 104 CFU/ mL) from stool samples that contain normal flora and inhibitors like semen samples. Moreover, other researchers have improved the sensitivity of the PCR using the DNA extracted from enrichment broth for PCR screening before Salmonella isolation; thus, the use of Chelex-100 and broth samples-should be considered in futures studies (Gwida and Al-Ashmawy 2014).
For avian coccidiosis, the results indicated that DNA from Eimeria spp. could be isolated by both methods and were detected by PCR for the first time in Argentina. Although the sensitivity of the analytic detection with M3 (9 tOP) was slightly higher than M1 (18 tOP). Kumar et al. (2013), have shown that samples containing between 50 and 100 tOP per reaction were 100 % positive for Eimeria genus PCR, while samples containing between 25-50, and <25 tOP protocol sensitivity was dropped to 80% and 60% positive, respectively, using a commercial kit, though. Thus, the detection of 18 tOP per reaction using M1 would be far enough for molecular diagnosis, and further field samples are being testing. It was shown that pre-cipitation with ammonium acetate followed by a resuspension with buffer Tris-EDTA after DNA extraction with Chelex-100 resin could improve its quality (Singh et al. 2018), demonstrating that this method would be enhanced.
On the other hand, the use of Chelex-100 has the advantage of not generating any adverse effect on the health of the operator in contrast with the use of organic solvents.
Finally, considering current values, the Chelex-100 method is 10 times cheaper than the commercial kit method, with costs per sample of $ 0.20 and $ 2.00, respectively. This represents a significant advantage for its application in veterinary diagnosis.
Even though the DNA extraction method for each pathogen must be tailored to the project objectives and the importance assigned to sensitivity, or other factors such as time consumption or human and financial resources, Chelex-100 could be considered as a feasible method for the development of molecular diagnostics in the veterinary field. Further studies with field samples are needed to determine the diagnostic sensitivity, and to validate this technique as a new diagnostic tool for equine paratyphoid and avian coccidiosis.
In conclusion, a simple and economic method was successfully employed for the first time to extract the DNA of Leptospira interrogans serovar Pomona Pomona, Brucella melitensis, Salmonella ser. Abortusequi, Leishmania spp., and Eimeria spp. for downstream applications, such as diagnostic PCR. This is especially important in low-resource settings in the veterinary field.

