











 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroducción
La candidiasis mucocutánea crónica es un trastorno raro y complejo, que se caracteriza por infecciones persistentes o recurrentes de la piel, las uñas y los tejidos de la mucosa, producida por Candida albicans en la mayoría de los casos.1Se observa principalmente en pacientes con síndrome poliglandular tipo I y generalmente se presenta antes que los signos y síntomas de las alteraciones endocrinológicas asociadas.2
Clásicamente, la presentación del síndrome poliglandular tipo I ocurre antes de los cinco años de edad y comienza con una candidiasis moderada pero recurrente como primer signo. Está principalmente localizado en las membranas mucosas orales, las uñas, el esófago y, más raramente, en los genitales y tracto gastrointestinal.3La infección fúngica de las uñas provoca decoloración y engrosamiento de la lámina ungueal, así como inflamación de la piel periungueal o paroniquia.4,5 La candidiasis oral tiene un pico de incidencia en los primeros dos años de vida y tiende a seguir un curso crónico.6 La presentación clínica de los pacientes con síndrome poliglandular tipo I también puede verse afectada por la aparición de hipoparatiroidismo. En esta condición, la piel es típicamente seca e hiperqueratósica, y las uñas son opacas y quebradizas con surcos longitudinales y alopecia.7
Reporte de caso
Niña de 15 años de edad con cuadro clínico de 12 años de evolución consistente en talla baja, dermatitis seborreica, alopecia difusa, con miniaturización del cabello, fotosensibilidad, micrognatia, paquioniquia e infecciones a repetición por Candida. Ha estado hospitalizada en varias oportunidades por cuadros respiratorios y hemorragia de vías digestivas altas.
Valorada por los servicios de Pediatría y Endocrinología, se le encuentra talla baja de 130 cm, peso de 23 kg y estadio 2 de la escala puberal de Tanner con botón mamario. El carpograma evidenció edad ósea de 10 años; la ecografía abdominal mostró genitales internos infantiles; radiografía de tórax sin alteraciones; resonancia magnética nuclear de hipófisis dentro de límites normales.
Los múltiples exámenes paraclínicos mostraron hipocalcemia, sin valores alterados de la hormona paratiroidea (PTH). Con todos estos hallazgos, se hace diagnóstico de talla baja patológica severa y déficit de hormona de crecimiento, por lo que se inicia suplemento hormonal para corregir esta alteración.
El cariotipo en todas las metafases analizadas mostró un complemento cromosómico normal 46XX. Los cambios dermatológicos de descamación en cuero cabelludo y en pabellones auriculares, además de placas eritematosas y descamativas en región malar, dorso de nariz, dorso de manos y tórax anterior, evidenciaron el diagnóstico de dermatitis seborreica. Asociado a esos hallazgos, se encontró que las uñas de las manos estaban hipertróficas e hiperpigmentadas. En el KOH de las uñas de las manos se encontraron micelios y macroconidias de Candida albicans.
El carpograma posterior al reemplazo hormonal mostró edad cronológica de 14 años. La niña presentaba placas alopécicas en cuero cabelludo, con miniaturización del cabello (figura 1). Tenía placas eritematosas y descamativas en rostro, tórax y en las cuatro extremidades, con bordes bien definidos y variedad de tamaño (figuras 2 y 3). Las uñas de ambas manos estaban engrosadas, amarillentas, distróficas y con eritema en los pliegues periungueales (Figura 4).
La paciente es la segunda de dos hermanos con los que comparte padre y madre. No se encontraron signos clínicos de hipoparatiroidismo ni de enfermedad de Addison al momento de la consulta.
Discusión
La presentación clínica de la candidiasis mucocutánea es muy variada. Esta diversidad clínica sugiere que bajo el denominador clínico antes descripto se agrupan diversas enfermedades específicas.8
Los síndromes poliglandulares autoinmunes (PGAS) o síndromes poliendocrinos autoinmunes comprenden un heterogéneo conjunto de enfermedades de aparición infrecuente y origen genético o espontáneo, que se caracterizan por la existencia de una respuesta de autoinmunidad contra varias glándulas endocrinas, aunque también pueden afectarse otros órganos. Comprende tres síndromes diferentes y otras enfermedades que tienen características parecidas. Los tres síndromes que se incluyen dentro de esta definición son el síndrome poliendocrino tipo 1 o APECED, el síndrome poliendocrino tipo 2 y el síndrome poliendocrino ligado al cromosoma X, también llamado XLAAD por las iniciales en inglés de las principales características:X-linked(ligado al cromosoma X), autoimmunity(autoinmunidad) yallergic dysregulation. Estos síndromes son distinguidos según la edad de presentación, patrones de combinaciones de enfermedades y diferentes modos de herencia.9,10 El síndrome poliglandular autoinmune tipo 1 es una enfermedad genética rara y compleja caracterizada por la disfunción hereditaria del sistema inmune, que afecta a múltiples sistemas de órganos que incluyen glándulas endocrinas y no endocrinas. El síndrome poliglandular tipo 1 se presenta como constelación de síntomas, que generalmente comienza en la infancia o la adolescencia y comúnmente involucra tres características: candidiasis mucocutánea crónica (CMC), hipoparatiroidismo e insuficiencia suprarrenal.3
El 60-80% de los pacientes con CMC manifiesta su enfermedad en la infancia, sólo una minoría la expresa tardíamente; en algunos casos, se comporta como una enfermedad familiar; predomina la herencia autosómica recesiva y es menos frecuente la herencia autosómica dominante. En una minoría de los casos se presenta en forma esporádica.11
Clínicamente, un individuo debe mostrar al menos dos de estas tres anormalidades para ser diagnosticadas como síndrome poliglandular tipo 1. El gen regulador autoinmune (AIRE) codifica un factor de transcripción que desempeña papel esencial en el mantenimiento de la tolerancia central. Se considera que las mutaciones AIRE forman la base genética del síndrome poliglandular tipo 1.12
Conclusiones
La incidencia de candidiasis sistémica está aumentando a una velocidad alarmante, especialmente en pacientes oncológicos y receptores de trasplantes con tratamientos inmunosupresores.13La presentación de síndrome poliglandular tipo 1 generalmente ocurre antes de los cinco años de edad y comienza con una candidiasis moderada pero recurrente como primer signo. Está principalmente localizado en las membranas mucosas orales, las uñas, el esófago y más raramente en los genitales y tracto gastrointestinal.14
Tradicionalmente, el tratamiento de los pacientes con candidiasis mucocutánea se basa en la terapia de las enfermedades infecciosas secundarias al déficit inmunológico y el tratamiento de las endocrinopatías y complicaciones asociadas, pero estos tratamientos son sólo paliativos: no resuelven el déficit inmunológico.15
El amplio espectro de manifestaciones del síndrome poliglandular tipo 1 es la base del tratamiento integrado. La estrategia terapéutica personalizada depende del órgano afectado. Por lo general, la terapia de reemplazo hormonal, la terapia antiinfecciosa y la terapia inmunosupresora son esenciales para el éxito del tratamiento.

Figura 1. Figura 2. Placas alopécicas y miniaturización del cabello Placas eritematosas y descamativas en el rostro
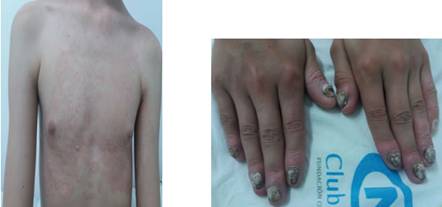
Figura 3. Figura 4 Placas eritematosas y descamativas en el tronco . Uñas engrosadas, distróficas y eritema en áreas periungueales


