











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCTION
Antiphospholipid syndrome (APS) is systemic autoimmune entity characterized by thrombotic phenomena (arterial and/or venous), fetal losses, and persistent increase in the serum level of an tiphospholipid antibodies.
Although the global prevalence is unknown, it is a rare condition, and it is estimated to be present in approximately 1 % of the general population. It can be primary or in the context of an underlying disease, usually systemic lupus erythematosus (SLE) or other systemic autoimmune disorders.
The most common pulmonary complications include: pulmonary thromboembolism, throm boembolic and non-thromboembolic pulmonary hypertension, microvascular thrombosis, acute respiratory distress syndrome, and alveolar hemor rhage, the latter being an uncommon and poten tially life-threatening manifestation.
CASE REPORT
A 16-year old male consults for a 1-year history of episodes of intermittent hemoptysis. Medical re cord: Primary APS, anticoagulated with acenocou marol for DVT and PTE, hospitalized for idiopathic thrombocytopenic purpura (ITP) (treated with gammaglobulin) and alveolar hemorrhage (AH) in February 2021, requiring invasive ventilation for 9 days. Reason for hospitalization: hemoptysis. Upon admission, the patient was hemodynamically stable; oxygen saturation at 98 %. Presumptive diagnosis of AH.
The following tests were performed: Labora tory: normal complement levels, negative ANCA antibodies, negative ANA (antinuclear antibodies), ESR (erythrocyte sedimentation rate) 23, negative anti-MPO antibodies, negative anti-PR3 antibod ies, negative anti-glomerular basement membrane antibodies. CHEST CT SCAN: Increased lung density with ground-glass opacity of diffuse pul monary infiltration predominantly peribroncho vascular in both lung fields. Consolidation area in the left base (Figure 1 and Figure 2). RFE + DIFUSSION: FVC 4.41 (81 %) FEV1 3.31 (73 %) FEV1/FVC 75 %. DLCO 141 % DL/AV: 186 % AV: 75 %. BRONCHOSCOPY: No endoluminal lesions. Bronchoalveolar lavage (BAL) in basal segments of the left lower lobe with negative cultures and cytology test showing 95 % siderophages. Oral an ticoagulation is discontinued, intravenous heparin is administered, cultures are taken, and antibiotic therapy is initiated. The patient responds favor ably and is then discharged.
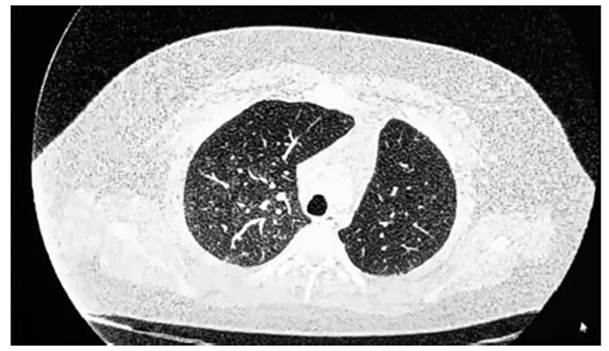
Fig. 1 Chest CT scan: diffuse bilateral ground-glass alveolar infiltrate, predominantly in the right lung field.

Fig. 2 Chest CT scan: ground-glass infiltrate of diffuse and predominantly peribronchovascular distribution in both lung fields. Consolidation in the left lower lobe (LII).
The diagnosis of AH in the context of APS is confirmed based on the CT scan infiltrates, the elevated DLCO (diffusing capacity of the lungs for carbon monoxide), and a high percentage of siderophages in the BAL. High-dose corticoste roid immunosuppression is initiated. No further episodes of hemoptysis.
DISCUSSION
The main form of pulmonary involvement in APS is pulmonary thromboembolism, with a frequency of 3.5-14.1 %. Chronic thromboembolic pulmonary hypertension (CTEPH) is a relatively rare compli cation. Poli et al reported the incidence of CTEPH after a first episode of pulmonary embolism (PE) to be 0.4 % in their series, which included 239 patients with PE.1
In a prospective long-term follow-up study, the cumulative incidence of CTEPH in patients with first-time diagnosed PE was found to be 11.2 % at 3 months, 12.7 % at 1 year, 13.4 % at 2 years, and 14.5 % at 3 years.2
In the study by S. Sarinc Ulasli et al, which included 67 patients, acute pulmonary throm boembolism (PTE) was detected in 11 patients (16.4 %), and alveolar hemorrhage in 2 (3 %). Four patients with acute PTE (36 %) developed chronic thromboembolic pulmonary hypertension. One patient developed CTEPH and diffuse alveolar hemorrhage after acute PE during follow-up.3
Alveolar hemorrhage (AH) is a rare and po tentially life-threatening condition in APS with a prevalence of less than 0.7 %. Hillerdal et al reported the first patient in 1991, and since then, approximately 100 cases have been published.4
In the series by Stoots et al, diffuse alveolar hemorrhage was the initial presentation of APS in 9/79 patients (11 %), and three out of 17 patients in a case series were diagnosed with APS only after presenting DAH. However, two other reviews of 18 and 13 patients observed a median onset of DAH of 5.9 and 5.8 years, respectively, after the diagnosis of APS. Additionally, many patients experienced diagnostic delay.5
The risk factors predisposing patients to alveo lar hemorrhage are not well known. Microvascular thrombosis and rupture of small pulmonary vessels are suggested as potential pathogenic mechanisms for alveolar hemorrhage in APS. It has been re ported that viral infections of the upper respiratory tract or bacterial pneumonia can trigger episodes of alveolar hemorrhage.6
The management of anticoagulation in APS patients with alveolar hemorrhage is complex, because discontinuing the anticoagulation entails a high risk of recurrent thrombosis. It is suggested to suspend anticoagulation during alveolar hem orrhage, to be restarted once it is under control.7
Corticosteroids induce remission in most pa tients; however, almost half of them experience recurrence and require a steroid-sparing im munosuppressant to maintain remission. Regimens based on cyclophosphamide or rituximab achieve the highest remission rates (50 %); other strategies include intravenous immunoglobulin, plasmapheresis, mycophenolate mofetil, and/or azathioprine.
The work of Cartin-Ceba et al states that no firm recommendations can be made for preferred immunosuppressive medications; cyclophospha mide or rituximab were the most commonly used immunosuppressive agents.8
CONCLUSIONS
AH is an rare presentation of APS. Bronchoscopy, BAL, DLCO, and chest CT are used to confirm the diagnosis and help discard other causes of alveolar hemorrhage. The pulmonary biopsy is the gold standard for confirming the diagnosis, although the histological pattern is not specific and is not routinely recommended.

