











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
PermalinkINTRODUCTION
Lambda interferons (IFNλ) are cytokines rapidly produced by most vertebrates during the innate immune response, constituting the first line of defense against viral infections (Lazear et al., 2015). IFNλ1, 2 and 3 were identified in 2003 (Kotenko et al., 2003; Sheppard et al., 2003) and in 2013 a functional form of IFNλ4 was firstly characterized (Prokunina-Olsson et al., 2013). The IFNλ4 locus (19q13.2) is highly polymorphic (Fang et al., 2020) and it was reported that some allelic variants can modulate the susceptibility, progression and response to treatments against different viral infections (Chatterjee, 2010; Bravo et al., 2014; Angulo et al., 2015; Ispirologlu et al., 2017; da Silva Cezar et al., 2020). Interestingly, the most favorable alleles in this regard correspond to mutations that are in strong linkage disequilibrium and restrict the expression, stability or antiviral activity of IFNλ4 (Booth and George, 2013; O’Brien et al., 2014; Prokunina-Olsson, 2019).
Throughout the evolutionary history of the genus Homo these mutations have suffered a positive selection pressure resulting in a differential global distribution which is correlated to the ancestry of different human populations and may affect the immune response to different pathogens (Key et al., 2014; Bamford et al., 2018). An intronic variant that reduces the antiviral activity of IFNλ4 (rs12979860, T˃C) was characterized as the main gene determinant of the response against Hepatitis C Virus (HCV). The rs12979860-T allele is associated with lower sustained virologic response (SVR) rates and a lower percentage of treatment success (Ge et al., 2009). On the other hand, the CC genotype was strongly associated with spontaneous resolution and lower susceptibility to HCV infection (Thomas et al., 2009; Pedergnana et al., 2012; Indolfi et al., 2014a; Fan et al., 2016). Moreover, genotyping of rs12979860 is recommended to predict the patient´s response to different antiviral treatments (Sharafi et al., 2012; Ramamurthy et al., 2018). Different correlations between rs12979860 and clinical phenotypes associated with other viral infections have also been reported, conditioning the susceptibility, evolution and/or response to treatment against Hepatitis B and D (Ispirologlu et al., 2017), Dengue (da Silva Cezar et al., 2020), HIV (Chatterjee, 2010; Zaidane et al., 2018), CMV (Bravo et al., 2014; Chmelova et al., 2019) and coronaviruses (Hamming et al., 2013).
The rs12979860-C allele has a global frequency of 0.23-0.55 in African populations; 0.53-0.80 for Europeans and 0.72-1.00 for Asians, with higher frequencies in eastern Asia. Data about the distribution of these variants in South American populations are scarce and tend to be biased due to the small sample size and the genetic admixture of the populations assessed. The Argentinean population´s ancestry is the result of a deep miscegenation, product of different migratory waves during the last centuries, which means that the European, Native American and African components (frequently underestimated) are present at different degrees in the gene pool of different cosmopolitan populations of the country (Avena et al., 2012). In this regard, the immunogenetic profiling of IFNλ4- rs12979860, and the association with its ancestry, may be a potential tool in both anthropological and biomedical studies associated with infectious diseases. The objective of this study was to determine the distribution of the allelic variants of rs12979860 in a cosmopolitan population of Buenos Aires, Argentina, whose ancestry had been previously determined by assessing a set of 106 biallelic SNPs (Ancestry Informative Markers) widely spaced and balanced throughout the genome, that can discriminate Native American, African and European ancestry (Avena et al., 2012).
MATERIALS AND METHODS
Study Design
This study comprised DNA samples from unrelated donors from both public and private hospitals blood banks in Buenos Aires, Argentina (n=96). Informed consent was obtained from all individual participants included in the study. Most of them (89/96) also agreed to provide information about the region/country of birth of all their grandparents, which was included in the data analysis. The study was approved by the Ethics Committee of the Hospital Italiano of Buenos Aires and was performed in accordance with the ethical standards adopted in the Declaration of Helsinki.
rs12979860 genotyping
Different genotypes of rs12979860 were determined by PCR-RFLP, as it was previously described (Sharafi et al., 2012). A 241 bp fragment was amplified by endpoint PCR (Taq Pegasus®, Productos Bio-Lógicos, Bs. As., Argentina) following a standard cycle (5 min at 94° C; 35 cycles of 20 s at 94° C, 20 s at 59° C and 20 s at 72° C; and 5 min at 72° C) and then digested with Bsh12361 restriction enzyme (Thermo Fisher, DE, USA; 1U/reaction) for 1 h at 37° C. The primers used were 5´GCGGAAGGAGCAGTTGCGCT3´ (Fw) and 5´TCTCCTCCCCAAGTCAGGCAACC3´ (Rv) and the resulting fragments (rs12979860-CC = 196 + 45 bp; rs12979860-CT = 241 + 196 + 45 bp; rs12979860-TT = 241 bp) were revealed by agarose gel electrophoresis (3%) stained with GelRed (Biotium, CA, USA).
Statistical analysis
The allelic frequencies were determined, and Hardy- Weinberg equilibrium was assessed using the chisquare test (Microsoft Excel GenAIEx 6.5, Peakall and Smouse, 2012) to compare the genotype distribution. Differences associated to European, Native American or African component were determined using T test (GraphPad Prism 9). In all statistical analysis a p<0.05 was considered as statistically significant and α=0.05 was set as the risk level.
RESULTS AND DISCUSSION
The average individual ancestry was estimated as 69.4% European, 26.3% Native American and 4.3% African. Frequencies lower than 0.02 were not included in the data analysis since they may be associated to technical artifacts. The European component was present in every tested sample, with individual frequencies ranging from 0.02 to 1. The Native American component was also detected but to a lesser extent, in 79% of the samples (frequencies 0.02-0.8). Finally, the African ancestry was detected in 41% of the samples, with a frequency range from 0.02 to 0.23 (Figure 1, modified from Avena et al., 2012). This evidences the multiplicity of origins of Buenos Aires´ population, resulting of the miscegenation between Native Americans, enslaved Africans who came mainly from West Africa and Mozambique until the first half of the 19th century (Fejerman et al., 2005) and European immigrants, mainly from Italy and Spain, who arrived in the country between 1870 and 1960 (Avena et al., 2006; Muzzio et al., 2018). These results are in line with previously published data (Avena et al., 2006), further challenging the European self-perception as Argentina’s identity.
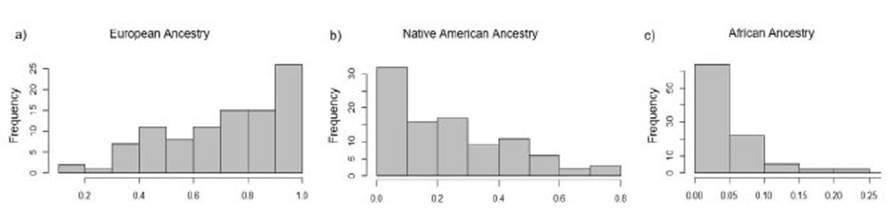
Figure 1 Frequency distribution of the individual European (a), Native American (b) and African ancestry (c) among healthy donors from Buenos Aires, Argentina, enrolled in this study (n=96). Modified from Avena et al. (2012).
Several studies have reported the distribution of the rs12979860 genotypes in different populations, mainly assessing its correlation with the susceptibility to different viral infections and response to antiviral treatments (Wu et al., 2012; Porto et al., 2015; Taheri et al., 2015; Echeverría et al., 2018). The correlation of this distribution and the local ancestry of these populations as well as its implications have also been assessed (Indolfi et al., 2014b; Rizzo et al., 2016), though this is the first report in an Argentinean global population. The overall distribution of rs12979860-CC, CT and TT was 29.17%, 50.0% and 20.83%, respectively. Hardy- Weinberg equation was used to calculate the genetic variation of this population at equilibrium. Significant differences were not detected (chi-square test: 0.00469; p=0.99766), thus suggesting that the impact of posible microevolutionary mechanisms and population structure is not significant. The allelic frequencies for C and T were 54.17% and 45.83%, respectively. These results differ from data reported in HCV chronically infected patients of a public center in Buenos Aires, with an allelic frequency of C=0.6 and 45.0% of heterozygosity (Machicote et al., 2018). This higher frequency of rs12979860-C is expected as it is known that this allele is favorable in both acute and chronic HCV infection. In this regard, the differences observed between healthy and infected individuals highlight the impact of assessing global populations when studying the distribution of this kind of markers.
A significant increase in the frequency of CC genotype was observed among donors with a strong European contribution (Figure 2a, p<0.05). Our results also suggest a greater impact of the Native American component among donors carrying the T allele (both CT and TT genotypes), although differences were marginally significant (Figure 2b). No differences in the rs12979860 distribution were directly attributable to the African component (Figure 2c, p>0.05), represented at low levels in our sample. Based on previously reported data on the composition and immigration patterns of the admixed population of Buenos Aires (Avena et al., 2012), we defined our parental population including sub-Saharan Africans (involved in slavery trafficking) and Europeans from Italy and Spain (Avena et al., 2006). To minimize bias, we only considered reported data on the rs12979860 distribution (Table S1) from non-cosmopolitan populations with a sample size greater than 50. Ethiopian Jews and Sephardic Jews from Rome, Italy, were also excluded, as these groups tend to be endogamous and have a different origin, which may introduce certain bias to our analysis. The mean frequency of the rs12979860-C allele for this parental population is 0.654 for Europeans, 0.298 for Africans and 0.518 for Native Americans (table S1). However, data available regarding the Native American component are scarce and are often based either on cosmopolitan admixed populations or studies with very small sample sizes and variable results. Despite the lack of a robust sample to perform comparisons, our results suggest that populations with greater autochthonous ancestry tend to exhibit higher frequencies of the rs12979860-T allele. Further studies are needed to fully characterize the distribution of this polymorphism in Latin America, as available data seem to be contradictory. To explain this it is important, regarding cosmopolitan populations, to disclose their composition and their genetic ancestry in order to determine their parental populations´contribution. Latin American cosmopolitan populations are known to be admixed, but the European, Native American and sub-Saharan contributions have marked regional differences. Hence the relevance of studying cases such as the one here described considering the genetic ancestry of the population under study.
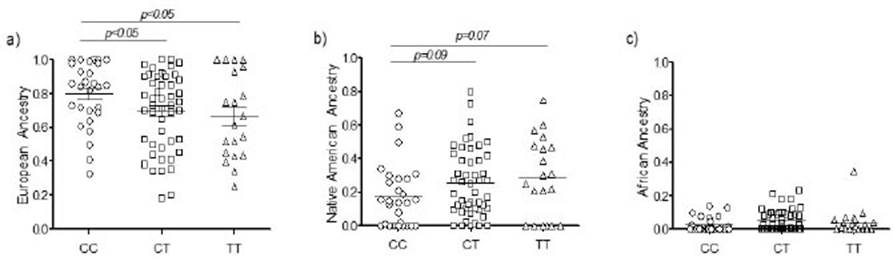
Figure 2 Distribution of European (a), Native American (b) and African ancestry (c) among individuals carrying rs12979860-CC, CT and TT genotypes. p<0.05 were considered as statistically significant.
The frequency of rs12979860-C in Buenos Aires´ individuals was similar to previously reported data for populations from Tuscany (C=0.603) in Italy, which are among the lowest compared to other West European populations (Table S1). The reported frequency of this allele in an Iberian population, however, was higher than the one described in our study (C=0.705, Table S1). Although immigrants from both Italy and Spain are the main determinants of the European ancestry of Buenos Aires´ population (Avena et al., 2006), it is to note that most of the immigrants in Buenos Aires (and Argentina) were of Italian origin (Avena et al. 2006). This may explain, at least partially, the frequencies here described. In order to further characterize the European contribution to the rs12979860 distribution we considered, when available, the self-reported data about grandparents’ origins. Interestingly, a total of 53 individuals declared the nonexistence of grandparents of European origin (8/53:CC, 33/53:CT and 12/53:TT), while only 36 individuals reported at least one grandparent from Italy, Spain/Portugal or other European countries (14/36:CC, 16/36:CT and 6/36:TT). This may be attributed to the fact that the vast majority of immigrants arrived in Buenos Aires before 1950. In our sample, the presence of Iberian ancestry seems to be underrepresented, as genealogical data suggest that the self-reported Italian ancestry was 33.0% higher than Iberian ancestry. Altogether, our results may be explained by the higher presence of Italian ancestry among European descendants in our sample, as well as by the admixture of these individuals with Native Americans and Africans or afro-descendants with a higher rs12979860-T frequency, thus increasing the heterozygosity and the rs12979860-T frequency. However, it is important to consider that, despite being very useful especially in regions with recent immigration patterns (Avena et al. 2012), this kind of surveys must be carefully analyzed, since different social and economic aspects may influence the individual self-perceived ancestry, as it was recently reported (Paschetta et al. 2021).
Notably, most of the populations that have been included in large-scale immunogenomic studies were of European origin, and might include certain bias by demographic, social and economic conditions of nonrandomly selected individuals (Peng et al., 2021). This may have affected the representativeness of the sample, thus compromising the conclusions of those studies. Therefore, increasing the genetic diversity while considering these structural inequalities is mandatory in order to obtain more reliable results. The PCR-RFLP protocol here applied was previously described and fully validated against PCR-sequencing, with a concordance of 100% in the results obtained for C/T alleles (Sharafi et al., 2012). In this regard, the use of a simple low-cost and high-yielding technique is paramount, since it allows small regional laboratories with limited resources to conduct population genetic studies, thus reducing the sampling bias that may occur in large cosmopolitan cities. This is particularly relevant in regions such as South America, in which the availability of qPCR or sequencing platforms is still limited. During the last years, there has been a growing interest on the impact of genetic ancestry on the immune response against viral infections (Mersha and Abebe, 2015). The molecular determinants responsible for those associations are being increasingly understood, and interferon pathways and their expression patterns seem to be influenced by genetic ancestry (Miretti and Beck, 2006; Randolph et al., 2021), as suggested by our results.
In the context of the COVID-19 pandemic and considering that IFNλ4 can elicit an antiviral response against RNA viruses, including some coronaviruses, several studies have assessed whether rs12979860 is involved in SARS-CoV-2 susceptibility and COVID-19 outcome. In this regard, it was reported that the T allele was overexpressed in COVID-19 patients compared to the general healthy population (36.2% vs. 26.4%), thus, this allele was proposed as a possible risk factor for COVID-19 (Saponi-Cortes et al., 2021). This was also supported by Rahimi et al. (2021), who demonstrated a positive correlation between the survival rate in COVID-19 patients and the rs12979860-CC genotype, which is also favorable to control other infectious diseases caused by RNA viruses. On the other hand, a higher frequency of the CC genotype among COVID-19 patients was reported in a different study, suggesting that people with the C allele (both CT or CC genotypes) are more susceptible to SARS-CoV-2 infection (Agwa et al., 2021). However, only slight differences between infected and control groups are shown (44.7% vs. 44.0%, respectively) and allelic frequencies are the same for both groups (C=34.0%, T=66.0%). In that study, it was also reported that 52.6% of the TT genotypes were classified as severe disease compared to 45.8% and 34.9% in the TC and CC genotypes, respectively (Agwa et al., 2021), which seem to be in line with the results published by Saponi-Cortes et al. (2021) and Rahimi et al. (2021). It is be noted, also, that the differences shown by Agwa et al. (2021) may not be exclusively explained by rs12979860 variants, considering that comorbidities were found in 57.4% of the infected group (and in 18.0% of controls). This highlights the relevance of carrying out a properly designed and unbiased sampling as well as a cautious analysis of the results in order to discern this type of controversies when assessing the differential distribution of these variants in different populations.
CONCLUSIONS
Given its importance and its apparent association with different infectious diseases, there is a growing interest in assessing IFNλ4 polymorphisms. As a whole, this study describes for the first time the distribution of rs12979860 polymorphism in a healthy sample of the population of Buenos Aires, Argentina, further demonstrating that these frequencies are associated to the composition of the population. This, in addition to being useful in anthropological studies, may contribute to the study of different infectious diseases for which interferon antiviral responses are key.

