











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkLa epilepsia es un trastorno neurológico cró nico no transmisible que abarca un espectro de disfunción cerebral que culmina en convulsio nes no provocadas y recurrentes. Las convulsio nes se manifiestan con movimiento de una parte o de todo el cuerpo y en ocasiones se acompa ñan de pérdida de consciencia. Las convulsiones se deben a descargas eléctricas excesivas en un grupo de células cerebrales, pudiendo ir desde episodios muy breves de ausencia a contraccio nes musculares prolongadas y graves. Hay que señalar, sin embargo, que sufrir una convulsión no significa que se padece epilepsia ya que hasta el 10% de las personas de todo el mundo tiene una convulsión a lo largo de la vida y solo tie nen un diagnóstico de epilepsia un 0,5%. La epi lepsia se define por dos o más convulsiones no provocadas repetidas, o la posibilidad de repetir la primera, en un periodo de tiempo (datos de la Organización Mundial de la Salud; OMS: https://www.who.int/es/news-room/fact-sheets/detail/epilepsy).
Se estima que casi 50 millones de personas en todo el mundo padecen epilepsia, con una pre valencia de 8.23 por 1000 personas, de las cuales 6 millones viven en Europa1. Según la OMS, la epilepsia es una de las principales causas de dis capacidad neurológica y mortalidad, con impor tantes costes económicos y sociales1. El impacto de la epilepsia en Europa ha sido reconocido en la Declaración de la Unión Europea sobre la Epi lepsia, en la que se pide a la Comisión y al Con sejo Europeos que den prioridad a esta enferme dad. La epilepsia es generalmente diagnosticada en niños, aunque la incidencia de la epilepsia aumenta con la edad, produciéndose la mitad de las nuevas crisis en personas de 65 años o más.
A pesar de la disponibilidad de más de 20 me dicamentos anticonvulsivos, aproximadamente el 36% de las personas con epilepsia vive con convulsiones no controladas2. Estas últimas son la principal causa de deterioro de la calidad de vida y de su capacidad para funcionar de forma independiente en las personas con epilepsia, con los costes asociados. El otro factor importante que compromete la calidad de vida y el trata miento eficaz de la epilepsia son las comorbili dades asociadas a la epilepsia (CAE). Definidas como distintas entradas clínicas durante la epi lepsia, las probabilidades de CAE en las personas con epilepsia son significativamente mayores en comparación con la población general. La prevalencia de las comorbilidades varía en función de los distintos tipos de epilepsia, y van desde una variedad de trastornos neurológicos y psiquiá tricos (ansiedad, depresión, trastornos por défi cit de atención, síndrome del espectro autista y psicosis) y neurocognitivos (déficit de aprendi zaje y memoria, bajo rendimiento académico y alteraciones de las funciones ejecutivas) hasta afecciones como migraña, trastornos del sueño y demencia. Las CAE pueden aparecer antes, con comitantemente o después del diagnóstico de epilepsia. Independientemente del momento de aparición, es probable que compartan factores contribuyentes comunes (genéticos, ambienta les, moleculares o estructurales) y mecanismos neurobiológicos con efectos perjudiciales sobre la causa clínica y el resultado terapéutico. Las CAE también imponen una carga significativa a las familias de las personas con epilepsia, ya que se relacionan con una respuesta deficiente a los fármacos antiepilépticos y una mayor mortali dad, lo que complica las intervenciones terapéuticas y la asistencia sanitaria.
Epilepsia congénita
Las crisis epilépticas neonatales se definen como un fenómeno electrofisiológico paroxís tico, caracterizado por la aparición transitoria de signos y síntomas debidos a una actividad neuronal anormal excesiva o sincrónica en el cerebro3. Según la definición tradicional, las cri sis neonatales se producen en los primeros 28 días tras el nacimiento de un neonato a térmi no o antes de las 44 semanas de edad gestacio nal en un neonato prematuro. Las convulsiones neonatales afectan a 1-3 neonatos por cada 1000 nacidos vivos. La mayoría de las convulsiones neonatales (50-80%) son no sintomáticas, y solo son detectables mediante electroencefalograma (EEG, convulsiones EEG o subclínicas)3. Hay que tener en cuenta que las convulsiones sintomáti cas agudas son la emergencia neurológica más común en los recién nacidos, siendo más fre cuentes en el periodo neonatal.
La vulnerabilidad del cerebro neonatal a pa decer crisis convulsivas es consecuencia de un estado de mayor excitabilidad en la corteza ce rebral. Esto se debe al funcionamiento inmaduro del receptor del neurotransmisor acido gamma-aminobutírico (GABA-A). En el cerebro maduro, el GABA induce la entrada de iones cloruro a la neurona y por consecuente, la hiperpolariza ción de la membrana neuronal tras la unión a su receptor, activando el cotransportador 2 de potasio-cloruro (KCC2). Esto reduce la excitabi lidad de la neurona, provocando que no pueda conducir el impulso. Los principales responsa bles son la alta expresión del cotransportador 2 de potasio-cloruro (KCC2) y la baja expresión del cotransportador 1 de sodio-potasio-cloruro (NKCC1) que caracterizan el cerebro maduro. En el cerebro inmaduro de los recién nacidos, sin embargo, la proporción de expresión del cotransportador es diferente debido a la mayor ex presión del NKCC1, por lo que cuando el GABA se une a su receptor específico, provoca la salida de iones cloruro de la neurona y, por lo tanto, aumenta la probabilidad de despolarización de la membrana celular neuronal. Es decir, el GABA actúa como un neurotransmisor excitador en el cerebro inmaduro neonatal. Esto implica una mayor excitabilidad de los circuitos de la corteza cerebral por la sensibilidad de las neuronas a la generación de potenciales de acción4. Hay que tener en cuenta que el cerebro de los niños está creciendo y desarrollándose y que, por lo tan to, la actividad convulsiva cambia a medida que crecen.
En nuestro laboratorio hemos investigado las consecuencias de la disminución de neuro nas inhibidoras de la corteza cerebral (que usan GABA como neurotransmisor) en un modelo de ratón con mutación en el gen Lis1 y demostra mos que existe un retardo en la maduración funcional del receptor del GABA-A, que incre menta su carácter excitador de las neuronas pi ramidales de la corteza cerebral5-7. Esto produce una hiperexcitabilidad local en el circuito y su giere que una leve displasia cortical, con anoma lía en la disposición de las neuronas, puede ser causa predisponente de epilepsia neonatal (Fi gura 1). Ya es conocido el carácter epileptogénico de displasias corticales focales, asociadas o no a síndromes genéticos.
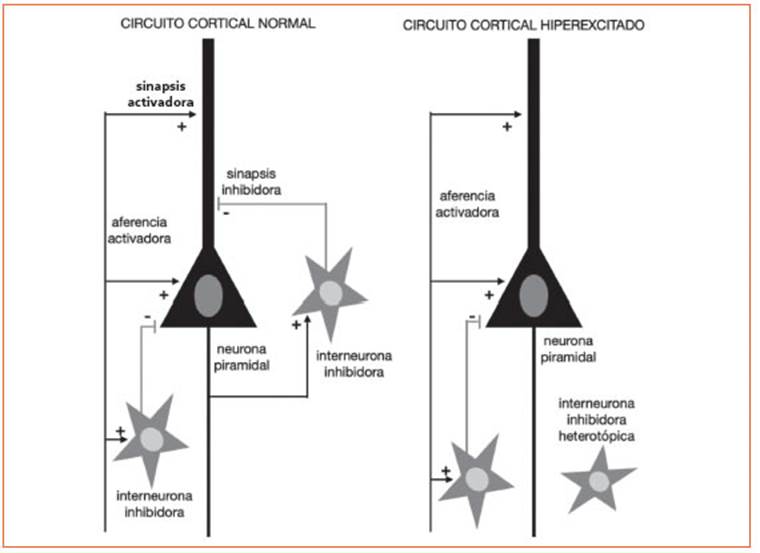
Figura 1 CIRCUITO NORMAL: Esquema simplificado del circuito cortical representando las entradas excitadoras al circuito (aferencias activadoras) que establecen sinapsis excitadoras (+) con las neuronas piramidales y con las interneuronas. Las colaterales axónicas de las células piramidales excitan a las interneuronas que envían axones que establecen sinapsis inhibidoras (-) con las neuronas piramidales. CIRCUITIO HIPEREXCITADO: En las displasias corticales las anomalías en la posición de las interneuronas inhibidoras (heterotopias neuronales) impide la formación de contactos de estas neuronas con las neuronas piramidales, lo que tiene como consecuencia la disminución de sinapsis inhibidoras y una mayor excitabilidad de las neuronas piramidales.
Como hemos visto, a un neonato se le puede diagnosticar epilepsia si ha tenido dos o más cri sis no provocadas, o después de una sola crisis si el niño muestra indicios de alta susceptibilidad a sufrir más crisis. La etiología depende de si el paciente es un neonato a término o prematuro. En el primer grupo, la causa más frecuente es la encefalopatía hipóxico-isquémica (EHI), y en el segundo, la hemorragia intracraneal (HIC)3,8. La causa de la epilepsia se conoce en el 54% de los niños, y en el 54% de estos se clasificó como como el síndrome epiléptico; en el 31% de las epilepsias congénitas se identifica una causa ge nética. Por lo tanto, en los niños pequeños con epilepsia, las pruebas genéticas deben ser prioritarias, ya que tienen el mayor rendimiento de cualquier otra prueba y es más probable que in formen también sobre la posibilidad de terapia de precisión y el pronóstico9. Tomando en con junto las epilepsias neonatales, se detectan al teraciones estructurales del cerebro en el 36% de los casos y en el 19% de los casos con diagnós tico genético. En cuanto a la evolución, a los dos años después de su presentación, el 36% de los niños presentaban epilepsia resistente a fárma cos (ERF) y el 49% retraso global del desarrollo (RGD). En epilepsia establecida, la causa se pue de determinar en el 82% de los casos de ERF y en el 75% de los de RGD. Puesto que las epilepsias de etiología indeterminada son más frecuentes en las comunidades desfavorecidas, existe pro bablemente un mayor riesgo multifactorial en estas poblaciones, donde factores ambientales pueden jugar un papel relevante en el desarro llo de la epilepsia (menos asistencia sanitaria al embarazo y al parto, mayor toxicidad ambiental y posibilidad de maltrato físico). La importancia del periodo perinatal en la reconfiguración del patrón de metilación de la cromatina puede im plicar una mayor sensibilidad del genoma a los procesos epigenéticos asociados a factores tóxi cos ambientales10.
La neuroinflamación es uno de los mecanis mos más comunes que subyacen a la epilep togénesis. Se han estudiado diversas vías neu roinflamatorias asociadas a una alteración de la permeabilidad de la barrera hematoencefálica y la producción de factores humorales proinfla matorios que modifican la actividad sináptica y favorecen la hiperexcitabilidad neuronal que, como hemos visto, esta aumentada en el cere bro neonatal. Vezzani y colaboradores11 descri ben que las principales causas neuropatológicas de las convulsiones neonatales son: anomalías estructurales, lesiones traumáticas, infeccio nes, isquemias y tumores. Todas estas causas están asociadas a procesos inflamatorios cere brales, que se verán potenciados por las crisis, si se repiten. El proceso consta de tres periodos: (1) la causa inicial produce una crisis; (2) que va seguida de un periodo latente; y (3) la fase de epilepsia establecida. Como hemos visto, la cri sis inicial puede deberse a lesiones neurológicas subyacentes o a causas desconocidas asociadas a hiperexcitabilidad del circuito cortical. A la fase inicial le sigue un periodo latente que implica el desarrollo de una neuroinflamación progresiva sin actividad convulsiva aparente. El periodo la tente puede durar días, semanas o meses y lue go progresa a la fase crónica, que se caracteriza por convulsiones recurrentes espontáneas junto con una neuroinflamación sostenida.
Comorbilidades en la epilepsia neonatal
La Real Academia Española de la Lengua defi ne como comorbilidad a la coexistencia de dos o más enfermedades en un mismo individuo, ge neralmente relacionadas. Aproximadamente el 50% de los adultos con epilepsia activa padecen al menos un trastorno médico comórbido (CAE). Varios estudios poblacionales a gran escala in forman de diversas afecciones clínicas que son hasta ocho veces más prevalentes en personas con epilepsia que en la población general. La im portancia de estas comorbilidades es cada vez mayor porque afectan al pronóstico de la epilep sia y a la calidad de vida. Por ejemplo, la migraña y las comorbilidades neurológicas y psiquiátri cas se asocian a un mal pronóstico de las crisis, mientras que la depresión se ha relacionado con una menor calidad de vida12.
Las comorbilidades neurológicas y psiquiá tricas son casi 5 veces mayores en niños y jó venes con epilepsia en comparación con la población general. La presencia de trastornos neurológicos y/o psiquiátricos que preceden a la aparición de las crisis en niños y jóvenes con epilepsia sugiere que hay alteraciones neuro biológicas subyacentes, que pueden ser causa de la predisposición a padecer epilepsia y de los procesos comórbidos de manera indepen diente. No obstante, las comorbilidades psi quiátricas en epilepsia infantiles y juveniles son generalmente multifactoriales, incluyendo anomalías estructurales, inmadurez cerebral y factores ambientales13.
Según Berg y colaboradores, la epilepsia se ha asociado a los siguientes procesos comórbidos14:
Alteraciones neurológicas: tumores cerebra les, trastornos del espectro autista, parálisis ce rebral, migraña y trastornos del sueño.
Enfermedades mentales: depresión, ansiedad y déficit de atención e hiperactividad.
Alteraciones cognitivas: discapacidad inte lectual, problemas de memoria y aprendizaje y disfunción ejecutiva.
Holmes15 indica que “muchas de las comor bilidades de la infancia tienen una asociación bidireccional, ya que la afección comórbida au menta el riesgo de epilepsia y la epilepsia aumenta el riesgo de la afección comórbida”. Esta característica bidireccional de la epilepsia y las CAE sugiere una base patológica subyacente co mún tanto para las crisis como para la condición comórbida (Figura 2). Aunque el reconocimiento de las CAE pediátricas es cada vez mayor, exis te probablemente un retraso en el desarrollo de terapias eficaces, en parte por la preocupación de que los fármacos utilizados para tratar las condiciones comórbidas pudieran aumentar la susceptibilidad a las crisis. Por ello este autor apunta la necesidad de ensayos clínicos aleato rizados y controlados con placebo sobre la efica cia y seguridad de los fármacos en el tratamien to de las comorbilidades de la epilepsia infantil.
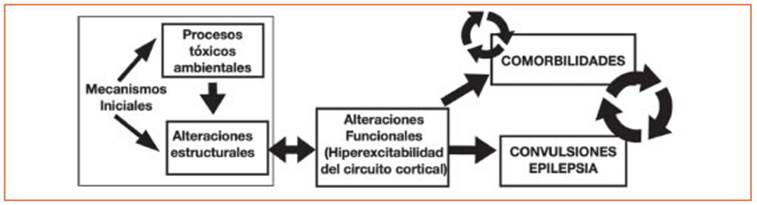
Figura 2 Esquema que representa las relaciones causales entre las causas subyacentes a las alteraciones funcionales de la epilepsia (hiperexcitabilidad del circuito cortical) que as su vez está implicado en el desarrollo de la epilepsia y las comorbilidades asociadas; que a su vez tienen relaciones predisponentes recíprocas entre ellas.
La relación bidireccional de comorbilidad entre epilepsia y autismo ha sido estudiada de forma muy intensa en una revisión sistemática de series de casos, donde se incluyeron 74 estu dios con 283.549 pacientes16. Los autores obser varon que la prevalencia media de la epilepsia en personas con autismo durante el periodo de estudio fue del 12,1% (en población general ya hemos visto que es del 0.8%), mientras que la prevalencia media del autismo en personas con epilepsia fue del 9.0% (en población general está entre 1.1 y 2%). Se probó que la prevalencia pe riódica de la epilepsia en personas con autismo, y viceversa, fue sistemáticamente superior a las estimaciones anteriores de la incidencia de es tos trastornos en la población general.
Se ha demostrado que la corteza cerebral de pacientes con autismo presenta un incremen to de microcolumnas corticales, con neuronas más pequeñas, hiperexcitabilidad intracolum nar y disminución de las conexiones largas de las neuronas corticales17. En una revisión siste mática que la displasia cortical y la hiperexcitabilidad microcolumnar de la corteza cerebral explican la relación entre autismo y epilepsia18.
La función normal de la corteza cerebral de pende del adecuado desarrollo y de la compleji dad estructural del tejido cerebral y, por lo tanto, son muchos los procesos que pueden alterar el equilibrio anatomo-funcional del cerebro y con llevar a la aparición de convulsiones que pue den evolucionar a epilepsia y/o el desarrollo del CAE19.
El diagnóstico precoz y el tratamiento de las comorbilidades psiquiátricas conducen a una mejora en la calidad de vida. Para ello se requie re un cribado rutinario, con biomarcadores ade cuados y un trabajo en equipo multidisciplinar.
Biomarcadores de epilepsia y comorbilidad
En los últimos años, los biomarcadores se han convertido en algo habitual en la investigación y la traslación clínica, y cada vez se reconoce más su valor como criterios de evolución en ensayos clínicos y estudios preclínicos. Los biomarca dores permiten una investigación más rigurosa de los mecanismos de la enfermedad, un mejor diagnóstico con estratificación de los pacientes, la comprensión de los procesos de la enferme dad y el seguimiento de la dinámica del trata miento para un control más eficaz.
En el campo de la epilepsia, las crisis con vulsivas se han definido históricamente como la única medida objetiva de la epileptogénesis de la enfermedad. Con el reconocimiento de biomarcadores ideales de la epilepsia, además de la patología primaria, se deberían abor dar las comorbilidades, incluido su diagnós tico precoz, predecir su gravedad, progresión y perspectivas, reflejar el mecanismo subya cente y proporcionar una orientación basada en datos para el tratamiento combinado con la epilepsia y el seguimiento de la eficacia del tratamiento.
Aunque hasta ahora no se ha identificado ningún biomarcador específico para las CAE, los avances en detección molecular, imagen cere bral, herramientas neurofisiológicas y otros mé todos prometen cambiar radicalmente la ges tión de esta compleja enfermedad. En el caso de la epileptogénesis, se están investigando como biomarcadores los cambios en el EEG, los datos de resonancia magnética funcional y estructural (MRI/fMRI), el análisis multiómico, los perfiles de miARN y los perfiles de proteínas específicas en biofluidos. Dado que las CAE pueden compartir mecanismos comunes con la epileptogénesis, debería considerarse su uso en la predicción, el diagnóstico y el seguimiento de epilepsia y sus comorbilidades. Aunque se están llevando a cabo investigaciones para identificar marca dores de comorbilidades específicas, se justifica la necesidad de un enfoque más integrado que permita un tratamiento multimodal guiado por biomarcadores objetivos y fiables para su diag nóstico y estratificación, que conduzca a inter venciones más eficaces.
Conflicto de Intereses: Los autores declaran que no existe con flicto de intereses.

