











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkLos schwannomas son los tumores benignos más frecuentes de los nervios periféricos. El schwannoma melanótico (SM) es una variante rara e infrecuente caracterizada por el depósito citoplasmático de melanosomas (melanina). A diferencia de las otras variantes de schwanno mas, tienen capacidad de malignización. El pri mero en describir esta entidad en un ganglio de la cadena simpática torácica fue Millar en 19321. Existen dos subtipos de SM: esporádico o no psammomatoso y psammomatoso. Este último puede formar parte de un trastorno genético au tosómico dominante (síndrome de Carney)2. Por poseer características y comportamiento distin tos al resto de los schwannomas, el SM fue recla sificado como “tumor maligno melanocítico de la vaina neural” en la 5ta edición de la clasifica ción de los tumores del sistema nervioso central de la Organización Mundial de la Salud en 20213.
Caso clínico 1
Mujer de 59 años, con antecedentes patológicos de artritis reumatoide, hipotiroidismo y cáncer de mama (cirugía conservadora de mama y posterior radiotera pia + tamoxifeno) fue evaluada en el Servicio de Cirugía Torácica por hallazgo de tumor de mediastino posterior en tomografía axial de tórax realizada durante un con trol post infección de COVID-19. En la misma se obser vó una formación nodular de 18.2 mm de bordes lisos y con densidad de partes blandas de probable localización extrapulmonar, ubicado por delante de la 2ª articulación costovertebral derecha, adyacente al segmento apical del lóbulo superior del pulmón derecho. Se le solicitó una re sonancia magnética de tórax para mayor categorización, en donde se informó una imagen nodular heterogénea de 20 mm con señal hiperintensa en T1, intermedia en T2, con restricción en la secuencia de difusión. Se ubica ba a nivel apical derecho, impresionaba ser extrapleural y podría corresponder a tumor de la vaina neural (Fig. 1A-C). Con la sospecha de tratarse de un tumor de mediastino posterior de origen neurogénico, se decidió su resección quirúrgica a través de un abordaje videotora coscópico. Durante la videotoracoscopia se confirmó la localización extrapleural del tumor, el cual no presen taba hiperpigmentación. Dado el riesgo de síndrome de Claude-Bernard-Horner por la ubicación del tumor, no se utilizó cauterio monopolar ni energía ultrasónica para la disección del límite posterior del mismo. Se realizó una resección completa y se envió a patología. El informe in traoperatorio informó una proliferación de probable es tirpe shwannica. El resultado definitivo de anatomía pa tológica fue: schwannoma melanótico (“tumor maligno melanocítico de la vaina neural”). Se realizaron técnicas de inmunomarcación sobre cortes de inclusión en pa rafina, efectuadas en equipo automático BenchMark-XT (Ventana-Roche) para las siguientes determinaciones: S100 positivo; Melan A positivo, HMB45 positivo; cromo granina negativo; sinaptofisina negativo; Ki67 2% (Fig. 2 A, B). Pese a los recaudos tomados, la paciente evolucionó con ptosis palpebral y miosis.
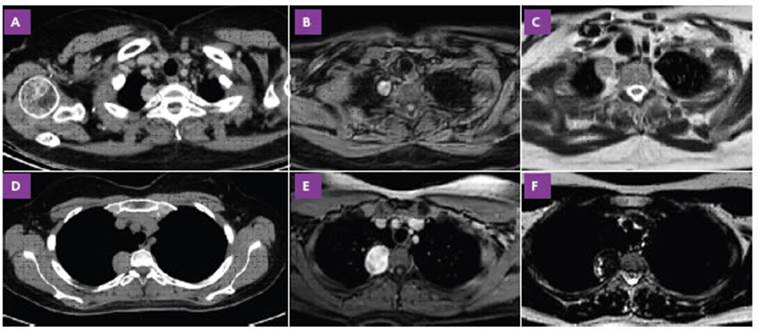
Figura 1 A-C (caso clínico 1). Tomografía axial computada de tórax (A): lesión en mediastino posterior de 18.2 mm de bordes lisos y con densidad de partes blandas (localización extrapulmonar). Resonancia magnética de tórax: imagen nodular heterogénea con señal hiperintensa en T1 (B), intermedia en T2 (C), con restricción en la secuencia de difusión, D-C (caso clínico 2). Tomografía axial computada de tórax (D): lesión nodular de 21 × 25 mm de bordes bien definidos en región apical derecha (paravertebral). Resonancia magnética de tórax: lesión redondeada ubicada en topografía paravertebral derecha, en íntimo contacto con el agujero de conjunción derecho (D3-D4) de aproximadamente 32 mm de diámetro máximo la cual posee comportamiento extrapleural, es hipointensa en secuencia T2 (F) e hiperintensa en T1 (E)
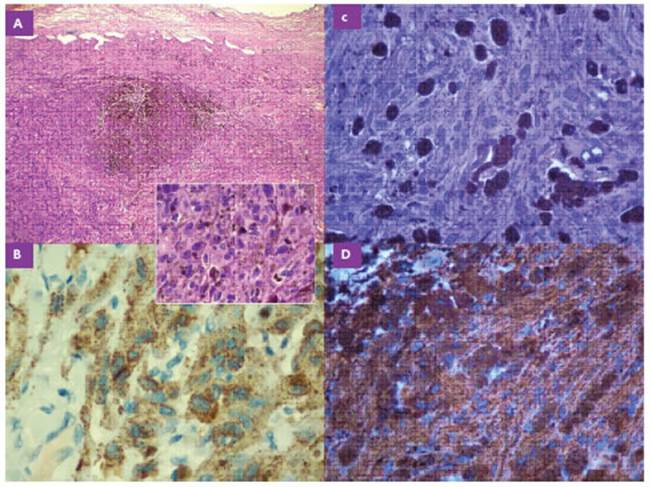
Figura 2 A-B: caso clínico 1. C-D: caso clínico 2. A: Hematoxilina-Eosina 4x: Proliferación neoplásica fusocelular y epitelioide bien delimitada con áreas pigmentadas. 40x: Celularidad neoplásica con moderado a marcado pleomorfismo, presencia de nucleolos evidentes y citoplasma amplio eosinófilo. Presencia de pigmento intracitoplasmático. B: Inmunohistoquímica. MELAN A: Positividad citoplasmática. C: Hematoxilina-Eosina 40x: Proliferación neoplásica fusocelular y epitelioide focal con moderado pleomorfismo y pigmento intracitoplasmático. D: Inmunohistoquímica. HMB 45: Positividad citoplasmática
Caso clínico 2
Mujer de 32 años, con antecedentes de enfermedad de von Willenbrand, con hallazgo de tumor en mediastino posterior en tomografía axial de tórax realizada en con texto de infección por COVID-19. En la misma se descri bió una imagen nodular de 21 × 25 mm de bordes bien definidos en región apical derecha (paravertebral) suge rente de tumor neurogénico. Fue controlada por su mé dico de referencia, quien le solicitó una nueva tomografía de tórax al mes y al decimosexto mes del hallazgo. En la última imagen se observó un aumento de tamaño a 25 × 33 mm. Se solicitó una resonancia magnética de tó rax para categorizar la imagen en donde se describía una imagen redondeada ubicada en topografía paravertebral derecha, en íntimo contacto con el agujero de conjunción derecho (D3-D4) de aproximadamente 32 mm de diáme tro máximo la cual poseía comportamiento extrapleural, era hipointensa en secuencia T2 e hiperintensa en T1 y con refuerzo post contraste (Fig. 1D-F). Con la sospecha de tratarse de un tumor de mediastino posterior de origen neurogénico, se decidió su resección quirúrgica a través de un abordaje videotoracoscópico. Durante la videoto racoscopia se confirmó la localización extrapleural del tumor, el cual presentaba una coloración negruzca con zonas de hemorragia. Se realizó una resección completa del mismo y se envió a patología para su estudio intrao peratorio (proliferación de origen neural). El resultado de finitivo de anatomía patológica fue: schwannoma mela nótico (“tumor maligno melanocítico de la vaina neural”). Se realizaron técnicas de inmunomarcación sobre cortes de inclusión en parafina, efectuadas en equipo automá tico BenchMark-XT (Ventana-Roche) para las siguientes determinaciones: S100 positivo; Melan A positivo, HMB45 positivo; CK negativo; Ki67 1% (Fig. 2C, D).
Una vez obtenido el diagnóstico definitivo ambos ca sos fueron discutidos de forma multidisciplinaria en el comité de oncología torácica del hospital por tratarse de entidades poco frecuentes y con un comportamiento más agresivo que los schwannomas. En los dos casos se deci dió no realizar ningún tipo de tratamiento adyuvante ya que las resecciones fueron completas (R0) y tenían una proliferación celular muy baja (Ki67 del 1 y 2%).
Al tratarse de dos reportes de casos aislados, el Comité de Revisión Institucional del Hospital Británico de Buenos Aires exime la presentación de consentimiento informa do para este formato de publicación. No se ha utilizado nombres, iniciales, número de historia clínica, u otros datos que permitan la identificación de los pacientes, en ninguna sección del reporte de los casos.
Discusión
Los schwannomas, neurinomas o neurilem momas son tumores que se originan de las célu las de Schwann siendo la variante más frecuente de los nervios periféricos, incluidos los nervios craneales. Por lo general son tumores benignos, únicos y de lento crecimiento. El schwannoma melanótico (SM) es un subtipo raro e infrecuen te caracterizada por el depósito citoplasmático de melanosomas (melanina). A diferencia de los otros subtipos de schwannomas, tienen capaci dad de malignización. Los porcentajes de malig nización y recurrencia descriptos en la literatura van del 9.1 al 42% y del 18.2 al 35%, respectiva mente. Una de las publicaciones más recientes y con mayor número de casos de SM (40) describen que los pacientes con un índice mitótico eleva do (≥2/10HPF) tienen mayor riesgo de maligniza ción por ser tumores con comportamiento más agresivo. Sin embargo, mencionan que un índice mitótico bajo no es sinónimo de benignidad, ya que el 50% de los pacientes con metástasis de la serie, no tenían un índice mitótico elevado.
Existen dos subtipos de SM: esporádico o no psammomatoso y psammomatoso. Este último puede formar parte de un trastorno genético au tosómico dominante denominado síndrome de Carney2, causado por mutaciones inactivadoras en el gen de la subunidad reguladora de la pro teína quinasa A tipo I-alfa (PRKAR1A). Este sín drome es una entidad infrecuente caracterizada por la presencia de lesiones pigmentadas en piel y mucosas, tumores mixomatosos (cardíacos y no cardíacos) y tumores endocrinos múltiples4,5. Los dos casos que presentamos se tratan de ca sos esporádicos de SM.
La localización más frecuente de los SM son las raíces nerviosas espinales dorsales6, la ca dena simpática1 y la médula espinal7. Las loca lizaciones extra espinales más frecuentes son el tracto gastrointestinal y los tejidos de partes blandas6. En la RMN, tienen un comportamien to opuesto a los schwannomas clásicos. Los schwannomas son hipointensos en secuencias T1 e hiperintensos en T2, mientras que los SM son hiperintensos en T1 e hipointensos en T28.
Histológicamente los schwannomas presen tan una arquitectura bifásica con patrones de organización celular denominados Antoni A y Antoni B. La región Antoni A es una zona hipercelular cuyas células son fusiformes con nú cleos que se disponen en empalizada formando filas paralelas y dando origen a los cuerpos de Veroca. La región Antoni B es una zona hipoce lular que se caracteriza por predominio de un estroma mixoide laxo con cambios degenerati vos. El análisis histológico y los marcadores de inmunohistoquímica son fundamentales para el diagnóstico definitivo de SM. En las células de Schwann de los SM se observan abundantes melanosomas citoplasmáticos (característica histológica fundamental que lo diferencia del schwannoma clásico) mientras que, en el resto, el pigmento marrón que se observa ocasional mente corresponde a lipofuscina y no a mela nina. Con respecto a la inmunohistoquimica, la positividad de la proteína S100 suele ser una ca racterística constante en los schwannomas. Sin embargo, en la bibliografía publicada encontramos que existen casos de SM con S100 negativa o moderada2,9. A diferencia de los schwanno mas clásicos, los SM tumores expresan mar cadores melanocíticos: HMB45, Melan-A y tirosinasa, por lo que el diagnóstico diferencial debería realizarse con tumores de localización mediastinal que expresen estos marcadores de inmunohistoquimica (ej.: melanoma)10. No obs tante, en la publicación de Lindholm y col. en donde presentaron 5 casos de SM mediastinal con presencia de depósitos intracelulares de melanina, ningún tumor expresaba marcado res melanocíticos11.
Al ser una entidad poco frecuente, no existen indicaciones precisas de adyuvancia una vez re secada la lesión. Entendemos que los factores pronósticos más importantes en cuanto a re currencia, diseminación a distancia y supervi vencia conocidos son: una resección quirúrgica completa y un índice mitótico bajo. Se necesitan más estudios para identificar factores de riesgo consistentes y para evaluar si debe considerar se tratamiento adyuvante en todos los casos, en casos seleccionados (resección incompleta - ín dice mitótico elevado) o únicamente luego una recurrencia o enfermedad metastásica. Al mo mento, la decisión de realizarla va a depender del análisis de cada caso en particular y de la experiencia de cada grupo tratante.

