










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.114 no.4 Buenos Aires ago. 2016
http://dx.doi.org/10.5546/aap.2016.e224
PRESENTACIÃN DE CASOS CLÃNICOS
http://dx.doi.org/10.5546/aap.2016.e224
SÃndrome de Melkersson-Rosenthal. Presentación de dos casos pediátricos
Melkersson Rosenthal syndrome. Report of two pediatric cases
Dr. Lucas Bordinoa, Dra. Mariana L. Juchlib, Dr. Marcelo Fernándezc, Dra. MarÃa de los Ãngeles Fitz Mauriceb, Dra. Andrea Martinsa, Dra. Zaida E. RamÃrezb y Dra. Roxana Spinia
a. Residencia de OtorrinolaringologÃa Infantil.
b. División de OtorrinolaringologÃa.
c. Servicio de OdontologÃa. Hospital General de Niños Pedro de Elizalde. Ciudad Autónoma de Buenos Aires.
Correspondencia: Dr. Lucas Bordino, lucasbordino2012@gmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 3-7-2015
Aceptado: 11-1-2016
RESUMEN
El sÃndrome de Melkersson-Rosenthal es una enfermedad muy poco frecuente en el paciente adulto y excepcional en la niñez y la adolescencia. El reciente trabajo publicado por G. Kayabasoglu et al., afirma que solo 30 pacientes se han descrito en la literatura internacional en edad pediátrica.
Es de etiologÃa desconocida, de curso crónico y progresivo, con compromiso neuromucocutáneo que afecta la inervación orofacial con infiltración granulomatosa no caseificante. ClÃnicamente, se caracteriza por una trÃada diagnóstica: edema recurrente de labios y/o cara, parálisis facial periférica aguda recurrente y alternante, y lengua escrotal o geográfica. Existen presentaciones oligosintomáticas y monosintomáticas.
Se describen dos pacientes de 7 y 11 años de edad, con presentación completa en un caso y oligosintomática en el otro. Consideramos tener en cuenta esta enfermedad infrecuente como diagnóstico diferencial ante la presencia de parálisis facial periférica aguda recurrente y/o edema de hemicara o labios por su comportamiento evolutivo.
Palabras clave: SÃndrome de Melkersson-Rosenthal; Parálisis facial; Recurrencia; Lengua geográfica.
ABSTRACT
Melkersson Rosenthal syndrome is a very infrequent disease of unknown etiology, chronic and progressive course, with neurocutaneous disease that affects the orofacial innervation and mucocutaneous tissues with non-caseating granulomatous infiltration. Clinically, it is characterized by the diagnostic triad: recurrent edema of lips and/or face; recurrent and alternating acute peripheral facial paralysis and scrotal or geographic tongue. The condition generally presents as oligosymptomatic or monosymptomatic form.
Two patients are presented with completed triad and oligosymptomatic form.
This recent review published by G. Kayabasoglu et al. states that only 30 patients have been described in the international literature in childhood. So we consider this disease in the differential diagnosis in the presence of acute peripheral facial paralysis and/or lips or facial edema.
Key words: Melkersson-Rosenthal syndrome; Facial paralysis; Recurrence; Geographic tongue.
INTRODUCCIÃN
Este sÃndrome fue descrito, en 1849, por Hubschmann y, en 1901, por Rossolino. En 1928, Melkersson reportó un caso de parálisis facial asociada a edema de labios. En 1931, Rosenthal agregó, como caracterÃstica adicional, la lengua con fisuras y resaltó el rol de los factores genéticos. En 1949, Luscher reunió estos signos y denominó el sÃndrome como sus dos autores.1
El sÃndrome de Melkersson-Rosenthal (SMR) es una enfermedad infrecuente de etiologÃa desconocida, de curso crónico y progresivo, con compromiso neuromucocutáneo que afecta la inervación orofacial.2 Es más común en mujeres y se manifiesta con mayor incidencia en la segunda década de la vida, sin predilección racial ni étnica.2-4
ClÃnicamente, se caracteriza por la trÃada que comprende edema de labios y/o cara con parálisis facial periférica aguda (PFPA) recurrentes y alternantes, y lengua escrotal o geográfica.2,3,5 Existen formas oligosintomáticas o monosintomáticas, en las que se presentan dos o un signo de la trÃada, respectivamente.
El diagnóstico es clÃnico cuando se presenta en forma completa.
Se describen dos pacientes de 7 y 11 años de edad, con presentación completa en un caso y oligosintomática en el otro.
Caso clÃnico 1
Paciente de 7 años de edad, de sexo masculino, que consultó por PFPA izquierda grado V de la clasificación de House Brackmann de 24 h de evolución (Figura 1), asociada a lengua geográfica con surco medio lingual profundo (Figura 2), seguido de edema de hemicara y labio homolateral, sin dolor ni otros sÃntomas acompañantes. El resto del examen fÃsico no presentaba particularidades.
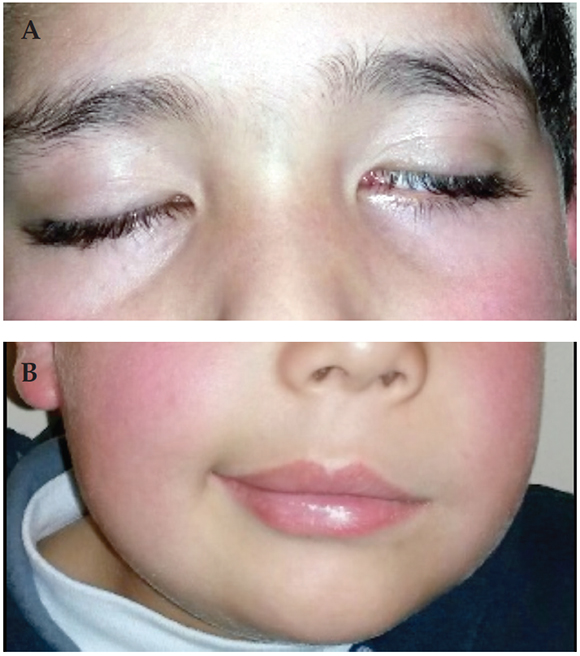
Figura 1. Parálisis facial izquierda. A: Signo de Bell. B: AsimetrÃa labial

Figura 2. Lengua geográfica con surco lingual profundo
ReferÃa, como único antecedente personal, haber padecido un cuadro clÃnico similar, del lado contralateral, 3 meses antes de la consulta, con resolución completa. Negó antecedentes familiares de importancia.
Caso clÃnico 2
Paciente de 11 años de edad, de sexo femenino, que se presentó a la consulta con PFPA izquierda grado Iv de la clasificación de House Brackmann, asociada a sÃndrome febril de 3 dÃas de evolución. Al momento del examen fÃsico, se constató la presencia de lengua geográfica. El resto del examen fÃsico no presentaba particularidades. ReferÃa, como único antecedente personal, haber padecido PFPA contralateral, de 15 dÃas de evolución con recuperación ad integrum, 2 años atrás. Negó antecedentes familiares de importancia.
En los dos casos, ante la sospecha de SMR y la necesidad de obtener un diagnóstico diferencial de otras etiologÃas, se solicitaron estudios de orina completa, hemograma, hepatograma, glucemia, uremia y dosaje de IgE, dentro de los lÃmites normales; las serologÃas IgM e IgG para virus del herpes simple I y II, varicela zóster, virus de Epstein-Barr, citomegalovirus fueron negativas. Prueba para virus de la inmunodeficiencia humana (vIH), negativa, y derivado proteico purificado (purified protein derivative; PPD, por sus siglas en inglés), no reactiva.
Se administró, por vÃa oral, 0,1 mg/kg/dÃa de betametasona en dosis decrecientes durante 10 dÃas, vitaminas del complejo B y, en forma empÃrica, 80 mg/kg/dÃa de aciclovir durante 7 dÃas. Se indicaron lágrimas artificiales, oclusión palpebral y rehabilitación kinésica.
Se interconsultó al Servicio de OftalmologÃa, que informó fondo de ojo normal, y al Servicio de NeurologÃa, con examen dentro de los parámetros normales. AsÃ, se descartaron causas infecciosas y sistémicas y se arribó al diagnóstico de SMR.
El edema facial presente en el caso 1 se resolvió en el trascurso de la semana de control. Ambos pacientes tuvieron recuperación completa de la parálisis facial a los 7 dÃas y a los 12 dÃas, respectivamente.
Ante la evolución clÃnica favorable de ambos pacientes, no se consideró necesario realizar estudios por imágenes.
El seguimiento continúa hasta el dÃa de la fecha en forma semestral o según demanda ante la aparición de nuevos sÃntomas, y no se registraron recurrencias en el lapso de un año en el primer caso y de cinco años en el segundo. Se han indicado a los padres las pautas de seguimiento a largo plazo por el carácter evolutivo de la enfermedad.
DISCUSIÃN
El SMR es inusual en niños. Su manifestación tiene un pico de incidencia entre la segunda y la tercera década de la vida.5,6
En dos trabajos recientemente publicados,5,7 se han descrito 30 casos en edad pediátrica en la literatura internacional. En el primero y en el segundo, se describe un seguimiento de pacientes con parálisis de Bell, en los que, durante su evolución, se constató que 5 de 49 niños sufrÃan SMR.
En Argentina, encontramos dos publicaciones que reportaban dos pacientes adultos con diagnóstico de SMR.3,4
Algunos autores describen igual afectación en ambos sexos; otros registraron mayor frecuencia en la mujer en relación de 3:1.2,6
El origen de la enfermedad es desconocido. Actualmente, tienen vigencia las teorÃas que vinculan el SMR con causas infecciosas por herpes simple tipo I y II, micobacterias, toxoplasma, espiroquetas2 y Mycobacterium tuberculosis; inmunológicas, desencadenado por alergia o intolerancia a alimentos; y genéticas.6,8,9
Histológicamente, se describen tres tipos de reacción: 1. Granuloma tuberculoide; 2. Granuloma sarcoidal; 3. Infiltrado linfonodular plasmático. Los granulomas son, en ocasiones, idénticos a los de la enfermadad de Crohn y la sarcoidosis. Se han encontrado granulomas en las paredes de los vasos linfáticos2 e infiltrado linfocitario perivascular, que afecta a los vasa vasorum y vasanervorum.10
El cuadro completo se manifiesta en el 25%-40% de los casos; las formas oligosintomáticas, monosintomáticas y secuenciales son las variantes más frecuentes de presentación,2,3,5,8 que representan el 40% de los casos en el comienzo de la enfermedad.
La manifestación más común de la forma monosintomática es la queilitis granulomatosa de Miescher (QGM), que se caracteriza por macroquelia aguda, preferentemente del labio superior, no indurada e indolora al inicio. Dura de horas a dÃas, con resolución completa en los episodios iniciales hasta hacerse finalmente persistente. El edema se va indurando y pueden aparecer grietas, ulceraciones y deformidad facial persistente y dolorosa.2,3,11,12 La lengua geográfica o escrotal y el surco medio lingual profundo se presentan en el 50%-70% de los casos; es el signo menos especÃfico, ya que se la observa frecuentemente en la población normal.6,11 La glositis granulomatosa es poco conocida; evoluciona hacia la macroglosia permanente y compromete las funciones del habla, la deglución y ocasiona alteraciones sensoriales en la lengua.5 Por último, la PFPA, que, generalmente, es unilateral y alternante, se presenta en el 30%-32% de los pacientes; puede preceder al edema por meses o años, desarrollarse tardÃamente, que es lo más común, o ser simultánea al edema. Con el tiempo, se hace permanente parcial o completa unilateral, que es lo más común, o bilateral y deja maloclusión palpebral y complicaciones oculares consecuentes, como la granulomatosis del párpado ipsilateral.13
Las recurrencias pueden presentarse en dÃas, semanas o años del cuadro clÃnico inicial.2,12 Pueden acompañarse de fiebre y sÃntomas generales leves.2
En el 50% de los pacientes, se hallan adenopatÃas regionales y, en algunos, adenopatÃas generalizadas. Pueden afectar otros nervios craneanos y dar sÃntomas neurológicos, como migrañas, neuritis, meningitis y polineuropatÃas periféricas.2,3,10,12
El diagnóstico se realiza sobre las caracterÃsticas clÃnicas y la exclusión de enfermedades sistémicas. En los casos mono- u oligosintomáticos que generen dudas, se debe realizar una biopsia de la región afectada para la confirmación anatomopatológica.2,5,12 En el diagnóstico diferencial, se deben considerar el angioedema, granulomatosis orofacial (Tabla 1) y las patologÃas que desarrollan lesiones granulomatosas, como la enfermedad de Crohn, la sarcoidosis, la tuberculosis y las micosis profundas.2-4,14
Tabla 1. Diagnóstico diferencial con granulomatosis orofacial

El SMR fue encontrado en pacientes con sÃndrome de Ehlers-Danlos, enfermedad de Crohn, rosácea y diabetes mellitus, respectivamente, los cuales son poco frecuentes de observar.2,9,14
Ambos pacientes presentados tuvieron los sÃntomas y antecedentes clÃnicos caracterÃsticos del SMR, coincidentes con las manifestaciones publicadas en la población adulta. Basándose en la presentación y evolución clÃnica y en los resultados de los exámenes complementarios, se descartaron enfermedades sistémicas en ambos. La realización de la biopsia no fue considerada en el edema del primer paciente, ya que manifestó la trÃada completa y su resolución completa y no generó duda diagnóstica en los sucesivos controles clÃnicos.
El objetivo del tratamiento por vÃa sistémica es disminuir la inflamación y paliar los sÃntomas. Primeramente, se utilizan los corticoides, con buenos resultados en la PFPA y el edema facial.6 Los inmunomoduladores, como la hidroxicloroquina, metronidazol, eritromicina, dapsona y anticuerpos monoclonales, tuvieron resultados poco favorables.15
Pueden utilizarse cortidoides intralesionales en la QGM.4
Se destacan el corto tiempo en que se resolvió en forma completa la PFPA en ambos pacientes (probablemente, esto se encuentre asociado a las primeras etapas de la enfermedad) y la rápida respuesta al tratamiento corticoideo.
CONCLUSIÃN
El SMR representa un desafÃo en cuanto a su diagnóstico y tratamiento. Esta enfermedad suele ser subdiagnosticada, por lo que hay que sospechar siempre esta patologÃa en pacientes que se presentan en forma mono- u oligosintomática. Dada su evolución crónica e irreversible, requiere un manejo diferencial por su seguimiento a largo plazo y sus recidivas. â–
1. Thiers BH, Miles DA, Roy S, Roger S. Trastornos que afectan la cavidad bucal. México: Interamericana; 1996.Págs.384-93. [ Links ]
2. Tucto Bautista S. SÃndrome de Melkersson-Rosenthal. Reporte de un caso. Folia Dermatol Peru 2004;15(3):176-82. [ Links ]
3. Nicola L, Oria F, Trila C, Abeldado A. Queilitis granulomatosa de Miescher. Dermatol Argent 2014;20(4):265-7. [ Links ]
4. Romano MF, Filice PA, Lauro MF, Peláez O, et al. Queilitis granulomatosa como parte del SÃndrome de Melkersson Rosenthal. Dermatol Argent 2014;20(5):339-43. [ Links ]
5. Kayabasoglu G, Yimaz MS, Fuat Varli A, Guven M. Melkersson-Rosenthal Syndrome in pediatric age group. Eur J Gen Med 2015;12(1):78-81. [ Links ]
6. Ziem PE, Pfrommer C, Goerdt S, Orfanos CE, et al. Melkersson-Rosenthal syndrome in childhood: a challenge in differential diagnosis and treatment. Br J Dermatol 2000;143(4):860-3. [ Links ]
7. Cosmelli Maturana R, Bravo Ahumada R, Cordova Jara L. SÃndrome de Melkersson Rosenthal: reporte de un caso y revisión de la literatura. Rev Esp Cirug Oral Maxilofac 2008;30(5):348-52. [ Links ]
8. Ruza Paz-Curbera E, Fernández BenÃtez M. Melkersson-Rosenthal syndrome in a diabetic boy. Allergol Immunopathol (Madr) 1998;26(6):291-3. [ Links ]
9. Darrouzet v, Houliat T, Lacher Fougere S, Bébéar JP. Paralysies faciales. EMC Oto-rhino-laryngologie 2002;20-260-A-10. [ Links ]
10. Camacho-Alonso F, Bermejo-Fenoll A, López-Jornet P. Queilitis granulomatosa de Miescher. Presentación de cinco casos. Med Oral Patol Oral Cir Bucal 2004;9(5):425-9. [ Links ]
11. Dodi I, Verri R, Brevi B, Bonetti L, et al. A monosymptomatic Melkersson-Rosenthal syndrome in an 8 years old boy. Acta Biomed 2006;77(1):20-3. [ Links ]
12. Goodhill V. Lesiones y heridas del VII par. En El oÃdo, enfermedades, sordera y vértigo. Barcelona: Salvat; 1986.Pág.594. [ Links ]
13. Palafox D, Vidal L, Tello-López B. SÃndrome de Melkersson-Rosenthal. Piel (Barc) 2013;28(9)558-60. [ Links ]
14. Cirpaciu D, Goanta CM, Cirpaciu MD. Recurrences of Bell's palsy. J Med Life 2014;7(Spec 3):68-77. [ Links ]
15. Simonsen AB, Deleuran M. Orofacial granulomatosis in children can be the initial manifestation of systemic disease: a presentation of two cases. Dermatol Reports 2014;6(1):5039. [ Links ]

