











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
El síndrome cérvico-óculo-acústico o de síndrome de Wildervanck (OMIM 314600) se caracteriza por la presencia de anomalía de Klippel-Feil (fusión de vértebras cervicales con vértebras en mariposa), síndrome de Duane (parálisis del VI par craneal con retracción del globo ocular en aducción) y sordera conductiva, neurosensorial o mixta.1 Además, se manifiestan otras características clínicas, tales como asimetría facial, paladar hendido, prominencias óseas en rama mandibular,2 anomalías vasculares diversas,3'4 malformaciones cardíacas5 y defectos en el desarrollo de la médula espinal.6
En relación con hallazgos radiológicos, se han descrito distintas malformaciones a nivel de oído medio e interno, como estrechamiento del conducto auditivo interno, malformación de Mondini y remanentes vestigiales de las estructuras de oído interno, así como ausencia del VI par craneal.4'6'7
Los primeros casos descritos datan de 1952 y hasta la fecha se han reportado 90 casos,8 de los cuales la mayoría son pacientes de sexo femenino, con una proporción 10:1. Debido a esto, se ha propuesto que tiene un modo de herencia ligado al X dominante, por lo que la ausencia de un gen funcional en estado hemicigoto podría causar una mayor tasa de letalidad en pacientes de sexo masculino que presentan el síndrome.4,6 No obstante, se proponen otros tipos de herencia, como la autosómica dominante y poligénica.7
En México, existe el reporte de un caso de un paciente de sexo masculino que presenta fusión de cuerpos vertebrales, hipoacusia grave bilateral y displasia coclear con canal auditivo interno estrecho. El cariotipo convencional que se le realizó no mostró alteraciones estructurales o numéricas en ninguno de los cromosomas.9
Al momento no existe un locus definido para el síndrome de Wildervanck, sin embargo, en un reporte reciente de un paciente de sexo masculino afectado con el síndrome, se descubrió mediante arreglo de hibridación genómica comparada (aCGH) una microdeleción de novo de 2 kb en la región Xp26.3, que escinde un único gen de esta región, el gen del factor de crecimiento de fibroblastos 13 o FGF13. Se especula que este gen participa en la embriogénesis, la morfogénesis, el crecimiento celular, la reparación de tejidos, la proliferación de progenitores neuronales, la migración neuronal y el crecimiento de conos. La pérdida o alteración de su estructura génica podría estar asociada a la falta de desarrollo del VI par craneal, el oído interno, la columna cervical, las extremidades, entre otras.6,10
El diagnóstico diferencial puede ser complejo, debido a la superposición de manifestaciones clínicas con otros síndromes más comunes, como los descritos anteriormente o con el síndrome de Goldenhar. Sin embargo, en este último, la microsomía hemifacial, las alteraciones oculares, junto con defectos vertebrales y la afección de ambos sexos en la misma proporción, nos orientan a su diagnóstico.11
Con el objetivo de describir las características clásicas del síndrome de Wildervanck, así como aportar nuevos datos al espectro fenotípico, se presenta el caso clínico de una paciente de sexo femenino que, además de tener la afección cervico-óculo-acústica, presenta malformación de extremidad superior izquierda y afección cardíaca no descrita con anterioridad.
CASO CLÍNICO
Paciente de sexo femenino de 14 años de edad, referida a la consulta de Genética con el diagnóstico de múltiples anomalías congénitas y cariotipo 46,XX,9qh+.
Antecedentes heredofamiliares negativos; producto de cuarto embarazo de padres no consanguíneos y sanos, embarazo normoevolutivo de 37 semanas, resuelto por vía vaginal. Al nacimiento, peso y talla dentro de parámetros normales.
Inició su padecimiento a los 4 meses de edad, con presencia de camptodactilia en mano izquierda, por lo cual fue intervenida quirúrgicamente en dos ocasiones. A los 4 años comenzó con limitación en los movimientos oculares de aducción bilaterales y estrabismo convergente; se le diagnosticó síndrome de Duane con ambliopía y astigmatismo hipermetrópico. A los 5 años manifestó datos de hipoacusia unilateral y a los 10 años de edad se evidenció escoliosis torácica.
A la exploración física, talla de 1,47 metros (pC 25), peso de 46,5 kg (pC 25), perímetro cefálico de 54 cm (pC 25). Cráneo normocéfalo, implantación capilar posterior baja, microsomía hemifacial izquierda, puente nasal ancho, ojos simétricos; ojo derecho con limitación grave a la abducción y aducción; ojo izquierdo con limitación grave a la abducción y leve a la aducción, enoftalmos leve en aducción de ojo derecho y moderado en el izquierdo. Cuello corto con limitación en arcos de movimiento, tórax corto, columna vertebral con escoliosis hacia la derecha, camptodactilia en mano izquierda. (Figura 1. A, B, C, D, E).
Se solicitaron los siguientes estudios de gabinete. Los potenciales evocados auditivos revelaron hipoacusia grave izquierda por lesión del receptor. Audiometría tonal, logoaudiometría y timpanometría con reporte de anacusia izquierda con ausencia de la función de reserva coclear y función disminuida del oído medio. En tomografía axial computarizada (TAC) de oído izquierdo, se observa malformación grave de oído interno, fusión de laberinto óseo con remanentes vestigiales de conductos semicirculares y cóclea (Figura 2. A). Ecocardiograma con reporte de prolapso leve anterior de válvula mitral. En resonancia magnética de columna, se observa anomalía de Klippel Feil; fusión de C4 a C5 con cuerpos vertebrales en mariposa que condiciona escoliosis cervicotorácica dextroconvexa (Figura 2. B y C). Ultrasonido renal sin alteraciones estructurales.
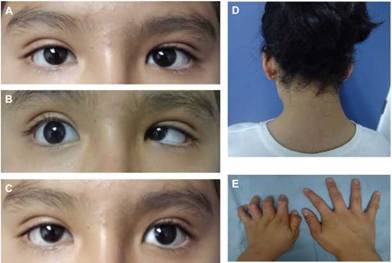
Figura 1: Síndrome de Duane tipo I. A) Mirada primaria con estrabismo. B) Mirada derecha con limitación a la abducción y enoftalmos moderado de ojo izquierdo. C) Mirada izquierda con limitación de la abducción, enoftalmos leve de ojo derecho. D) Implantación capilar posterior baja. E) Camptodactilia en mano izquierda
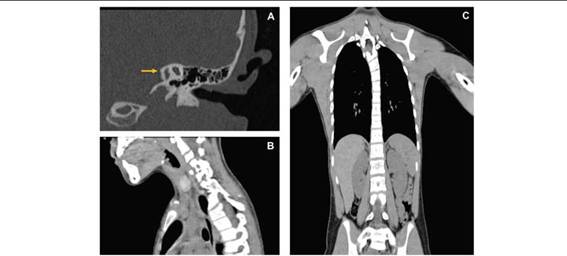
Figura 2: A) Tomografía axial computarizada de oído izquierdo. La flecha indica los remanentes vestigiales de cóclea, vestíbulo y conductos semicirculares. B) Resonancia magnética de columna cervical (corte axial): fusión de vertebras a nivel de C4-C5. C) Resonancia magnética de columna (corte coronal): elevación escapular, anomalías de segmentación a nivel cervicotorácico y deformidad de cuerpos vertebrales
DISCUSIÓN
El síndrome de Wildervanck es una enfermedad genética rara, caracterizado por una tríada fenotípica que incluye la anomalía de Klippel-Feil, el síndrome de Duane y sordera congénita.12 Nosotros presentamos un caso de una paciente de sexo femenino que cumple con los criterios diagnósticos del síndrome y que, además, presenta camptodactilia en mano izquierda, así como una alteración estructural del corazón, caracterizada por prolapso leve de la válvula mitral; estas últimas no descritas con anterioridad en otros pacientes, por lo cual, podrían considerarse dentro de la expresividad variable de la anomalía de Klippel-Feil.13 Al igual que otros casos esporádicos reportados en la literatura, en este caso no se encontró ningún antecedente familiar ni datos de consanguinidad.
Debido a que los pacientes con síndrome de Wildervanck presentan variedad de dismorfias y alteraciones viscerales, el cariotipo convencional es el primer estudio diagnóstico solicitado para evaluar la constitución cromosómica, como se ha reportado en varios pacientes incluida la nuestra.9'14
Hasta la fecha, no se ha esclarecido una etiología específica para el síndrome de Wildervanck, sin embargo, se han propuesto algunos genes candidatos, como FGF13, que interviene en los procesos de proliferación y migración neuronal,6 o los genes del factor de diferenciación de crecimiento 3 y 6 (GDF3, GDF6), en los cuales se han descrito mutaciones asociadas al síndrome de Klippel Feil 1, que intervienen en el desarrollo de malformaciones articulares y cardíacas.13 Los genes FGF3 y HOXA 1 están asociados con aplasia laberíntica, que se produce durante la tercera semana de gestación, sin embargo, también se relacionan con otros síndromes, como el de aplasia laberíntica, microtia y microdoncia (LAMM), y síndrome de mutación HOXA1, caracterizado por aplasia de la carótida bilateral, retraso en el crecimiento y alteraciones oculares.15 Lo anterior podría indicar en el caso de nuestra paciente un tipo de herencia poligénica, dada la afección a nivel articular y cardíaco en conjunto con las características clínicas clásicas del síndrome.
Será recomendable considerar la realización de pruebas diagnósticas basadas en citogenética molecular o secuenciación, debido a los antecedentes publicados en la literatura, que muestran alteración en genes candidatos, en FGF13 y microdeleción en Xp26.3.6'10
El tratamiento de estos pacientes incluye la realización de procedimientos quirúrgicos en casos de alteraciones estructurales que comprometan la función y terapia física para reducir la limitación por los defectos óseos; así como un seguimiento multidisciplinario, dependiendo de las necesidades de cada paciente, para guiarlo en el autocuidado.12
CONCLUSIONES
En los pacientes con anomalía de Klippel-Feil, es importante indagar de manera intencionada alteraciones musculoesqueléticas adicionales, malformaciones cardíacas, así como realizar evaluación audiológica y oftalmológica para descartar el síndrome de Wildervanck. La integración de la tríada clásica del síndrome permite el diagnóstico oportuno y favorece la intervención temprana, que tendrá impacto en la calidad de vida de los pacientes.
Recibido: 28-2-2022.
Aceptado: 21-7-2022.

