











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El hipotiroidismo congénito (HC) se describió por primera vez hace varias décadas, como una enfermedad en niños que presenta características de hipotiroidismo y, en ocasiones, causa discapacidad intelectual grave y retraso del crecimiento.1 En recién nacidos, su forma más común es el HC primario, con una prevalencia aproximada de 1:2000 a 1:4000. Surge por defectos del desarrollo de la glándula tiroides, conocida como disgenesia tiroidea, o por alteraciones en la biosíntesis de la hormona tiroidea, conocida como dishormonogénesis tiroidea.2 En el 80% de los casos existe un defecto estructural de la glándula tiroides, mientras que en el 20% restante se identifica glándula tiroides normal o agrandada; sin embargo, se presentan defectos en la síntesis de la hormona tiroidea. Se han reportado también formas leves de HC que se producen como resultado de un aumento transitorio o permanente de los niveles de la hormona estimulante de la tiroides (TSH) y se ha denominado "hipotiroidismo subclínico", si bien, para algunos autores, sería más adecuado denominarlo como "hipertirotropinemia".3 El propósito de esta revisión es resumir el conocimiento actual sobre las bases moleculares del HC con presencia de disgenesia tiroidea o dishormonogénesis, así como proporcionar una breve actualización sobre su diagnóstico y tratamiento.
Métodos
Se sintetiza la información actualizada en una revisión sobre las bases moleculares del HC, abordando la epidemiología, los principales genes de susceptibilidad, los estudios de asociación de genoma completo (GWAS, genome-wide association studies), los polimorfismos de nucleótido único (SNP, single nucleotide polymorphism), el diagnóstico molecular y el tratamiento. Se realizó una revisión bibliográfica narrativa en las siguientes bases de datos: Medline/PubMed, LILACS-BIREME y SciELO. Se llevó a cabo una búsqueda avanzada utilizando los operadores AND y OR, utilizando las siguientes palabras clave: hipotiroidismo congénito (congenital hypothyroidism), genética (genetic), polimorfismos SNP (polymorphisms SNPs). Como criterios de inclusión se consideraron los artículos de texto completo (en idioma inglés o español), los trabajos originales (cualitativos, cuantitativos o mixtos) y las revisiones bibliográficas; se excluyeron las cartas al director/editor y los textos de opinión.
Resultados
Una vez implementadas las estrategias de búsqueda y de recopilar la información, en el período 2000 a 16 de agosto de 2020 y aplicando los criterios de inclusión y exclusión, se obtuvieron 927 artículos. Se seleccionaron para revisión del texto completo 58 trabajos, en los que se consideraron el contenido de mayor relevancia para describir el tema abordado y el acceso al texto completo del artículo.
Bases genéticas del hipotiroidismo congénito
El HC es la pérdida parcial o completa de la función de la glándula tiroides, que afecta a los lactantes desde el nacimiento y da como resultado un deterioro grave del neurodesarrollo si o es tratado; es uno de los trastornos endocrinos congénitos más comunes, con una prevalencia de 1 por cada 3000 a 4000 recién nacidos.4 Las causas de HC se pueden clasificar en dos grupos: defectos del desarrollo de la tiroides (disgenesia tiroidea) y errores innatos de la biosíntesis de la hormona tiroidea (dishormonogénesis). Se han descrito al menos 12 genes que codifican para proteínas que, al producirse mutaciones, están implicados en el HC.
Genes asociados con hipotiroidismo congénito con disgenesia tiroidea
Entre el 80% y el 90% de los casos de HC es causado por disgenesia tiroidea, la cual presenta alteraciones en la morfogénesis de la glándula tiroides. Mediante la diferenciación por gammagrafía y ecografía tiroideas se pueden clasificar en: agenesias o atireosis; hipoplasia, cuando la tiroides es inferior al tamaño normal y con una ubicación anatómica usual, y ectopia, cuando la glándula tiroides, generalmente hipoplásica, está situada fuera de su sitio normal.5 La patogenia se desconoce en gran medida, sin embargo, se han sugerido la posible participación de factores ambientales, genéticos y epigenéticos; asimismo, se han asociado con la enfermedad ciertas mutaciones en los genes PAX8, NKX2-5, FOXE1, NKX2-1, GLIS3 y TSHR. A continuación, se describe lo observado en las distintas mutaciones.
PAX8
Codifica para un factor de transcripción de dominios emparejados y se expresa en la tiroides en desarrollo, los riñones y varias áreas del sistema nervioso central. Al unirse a regiones promotoras a través de sus 128 aminoácidos emparejados, regula la expresión de genes que codifican para producir tiroglobulina y peroxidasa tiroidea. Se han identificado varias mutaciones del gen PAX8 en pacientes con HC.6 La mayoría de estas mutaciones han causado disgenesia tiroidea; sin embargo, algunos pacientes con la mutación PAX8 tienen una glándula tiroides de tamaño normal.7
NKX2-5
Se expresa en el desarrollo de la tiroides y el corazón, lo que sugiere que este factor de transcripción regula rasgos de desarrollo comunes en los dos primordios de órganos.8 Mediante ensayos en animales de experimentación, se ha observado en embriones de ratones que, en ausencia de NKX2-5, se produce hipoplasia tiroidea además de defectos cardíacos.9 Se han detectado varias mutaciones de NKX2-5 en pacientes con ectopia tiroidea o atireosis.8 Por otra parte, se han identificado mutaciones de NKX2-5 en pacientes con una variedad de otras anomalías cardíacas, por lo tanto, este gen puede ser uno de los factores asociados con la prevalencia de malformaciones cardíacas en niños con HC.10
FOXE1
Codifica para el factor de transcripción tiroideo 2, que regula la transcripción de tiroglobulina y tiroperoxidasa. Estudios en embriones de ratones con ausencia de este gen, desarrollan paladar hendido y atireosis o una glándula sublingual ectópica.7 Hasta ahora, solo se han identificado tres mutaciones del gen en los seres humanos, todas homocigotas y en familias consanguíneas. La mutación usualmente está localizada en el dominio de unión al ácido desoxirribonucleico (ADN) del gen, lo que ocasiona que se codifique un FOXE1 defectuoso que tiene una actividad transcripcional insignificante.11 Al ser las mutaciones del FOXE1 inusuales, se recomendaría restringir el análisis de secuenciación de esta mutación a pacientes con al menos tres elementos del síndrome de Bamforth-Lazarus, debido a que en individuos en los que se ha reportado presentan únicamente HC, los resultados obtenidos han sido negativos.12
NKX2-1
Codifica para un factor de transcripción que se ha observado se encuentra implicado en el desarrollo de la glándula tiroides y de otros órganos.13 Su papel en la fisiopatología del HC fue sugerido al observarse en pacientes que tenían deleciones cromosómicas que abarcaban el locus NKX2-1.14 Estudios posteriores encontraron mutaciones puntuales en el gen que confirmaron su implicación en el fenotipo que incluye HC y síndrome de dificultad respiratoria.15,16 La mayoría de las mutaciones en el gen NKX2-1 ocurrende novo, sin embargo, se han comunicado algunos casos de transmisión dominante.17
GLIS3
Gen en el que mutaciones se han asociado con pacientes con HC y diabetes neonatal permanente, junto con restricción del crecimiento intrauterino, glaucoma congénito, fibrosis hepática y riñones poliquísticos.18 Un estudio llevado a cabo en una familia consanguínea de Arabia Saudita mostró que el gen GLIS3 presentó una inserción homocigótica en el afectado, lo que provocó un cambio de marco y una proteína truncada. En otro estudio de dos familias consanguíneas, una de Arabia Saudita y la otra gitana francesa, se encontraron distintas deleciones homocigotas en GLIS3, con un fenotipo tiroideo aparente de atireosis.
TSHR
Gen que presenta ciertas mutaciones que pueden provocar la inactivación del receptor de TSH;19 esto fue reportado por primera vez en 1995, observándose hipertirotropinemia asintomática, con una glándula tiroides de tamaño y captación normal de radioyodo.20 Varias mutaciones del gen TSHR inactivante se han informado desde entonces. Se observa que los heterocigotos son estrictamente normales o pueden tener una elevación muy leve de la TSH plasmática.21 El fenotipo de los homocigotos y heterocigotos compuestos es muy variable, desde hipertirotropinemia asintomática hasta HC grave con atireosis aparente. Por lo tanto, se debe considerar la secuenciación de TSHR en pacientes con los fenotipos descritos antes, especialmente si existe consanguinidad de los padres o antecedentes familiares que sugieran transmisión autosómica recesiva.
Genes asociados con hipotiroidismo congénito con dishormonogénesis
Varios genes se han relacionado con defectos en la biosíntesis de hormonas tiroideas, conocido como dishormonogénesis, lo que constituye un grupo de errores congénitos que causan el bloqueo total o parcial de los procesos bioquímicos implicados en la síntesis y secreción de hormonas tiroideas. De la etiología global, la dishormonogénesis representa del 10% al 20%.5 Los genes que se han visto asociados se mencionan a continuación.
TPO
Codifica para la peroxidasa tiroidea, que es una enzima unida a la membrana que participa en la biosíntesis de las hormonas tiroideas.22 Se han descrito mutaciones de TPO en diversos grupos étnicos. Hasta ahora, se han identificado más de 60 mutaciones inactivadoras relacionadas con el gen TPO, incluidas mutaciones, errores de empalme, deleciones e inserciones de nucleótidos. Dichas mutaciones son las principales responsables de la dishormonogénesis tiroidea, y constituye uno de los defectos hereditarios más frecuentes en el HC.23 Están presentes en prácticamente todos los pacientes con defectos permanentes de organificación del yodo total (TIOD, total iodine organification defect). Aunque, las mutaciones heterocigotas de TPO no provocan directamente una función tiroidea anormal, estos defectos monoalélicos pueden desempeñar un papel como factor de susceptibilidad genética en el hipotiroidismo transitorio.
DUOX2
Codifica una de las oxidasas duales que se encuentra en la membrana apical de los tirocitos y genera el peróxido de hidrógeno que necesita la peroxidasa tiroidea para la incorporación de yodo a la tiroglobulina,25 un paso esencial en la síntesis de la hormona tiroidea. Las mutaciones en el gen DUOX2 son una causa de HC y ha sido descrito en varios estudios.26 Aunque la mayoría de los casos de dishormonogénesis tiroidea se heredan de manera autosómica recesiva, las mutaciones monoalélicas o múltiples de DUOX2 también pueden conducir a HC debido a la deficiencia de H2O2.27 Se ha descrito que alrededor del 14.9% de los casos de HC con dishormonogénesis podrían deberse a mutaciones en el gen DUOX2.28
IYD
Gen que codifica para una enzima involucrada, junto con NADPH, en la desyodación reductora de la monoyodotirosina y la diyodotirosina, que son los subproductos de la producción de hormona tiroidea que conducen a la formación de yoduro libre y tirosina, los cuales pueden reutilizarse en la síntesis de hormonas.29 La enzima se expresa en el tirocito, el hígado y el riñón. Se han descrito mutaciones en IYD en varias investigaciones; en estudios in vitro las mutaciones bloquean la capacidad de IYD para deshalogenar la monoyodotirosina y la diyodotirosina.30 La pérdida de actividad de IYD evita el "reciclaje" normal de yoduro intratiroideo y conduce a una secreción urinaria excesiva de monoyodotirosina y diyodotirosina. Dado que la deficiencia de yoduro resultante puede no manifestarse al nacer, los pacientes notificados con mutaciones bialélicas de IYD pueden considerarse normales en el cribado neonatal de HC.29 Usualmente, estos casos acuden posteriormente a atención médica entre los 1.5 y los 8 años por las secuelas del hipotiroidismo no tratado.31 El hallazgo de mutaciones del gen TPO en un recién nacido con HC indica que el paciente requerirá un tratamiento de por vida con hormona tiroidea.24
TG
El gen codifica para producir una proteína llamada tiroglobulina, una de las proteínas más grandes del cuerpo.32 Esta proteína se encuentra solo en la glándula tiroides; la tiroglobulina se combina con el yodo y se modifica para liberar pequeñas moléculas conocidas como hormonas tiroideas.33 Las hormonas tiroideas desempeñan un papel importante en la regulación del crecimiento, el desarrollo del cerebro y la tasa de reacciones químicas en el metabolismo.
El estudio del TG es de gran relevancia para la fisiopatología tiroidea. Se han informado mutaciones del gen TG humano y se han asociado con HC34y bocio simple. El TG también ha sido identificado como el principal gen de susceptibilidad para la enfermedad tiroidea autoinmune familiar (AITD, autoimmune thyroid disease), mediante análisis de ligamiento utilizando marcadores de polimorfismos.35 La prevalencia de pacientes con mutaciones del gen TG es de aproximadamente 1 de cada 100 000 nacidos vivos. Se han descubierto cerca de cincuenta mutaciones en el gen TG humano.
SLC26A4
Gen que codifica para una proteína llamada pendrina, la cual transporta iones cloruro, yoduro y bicarbonato a través de las membranas celulares.36 La pendrina se produce en varios órganos y tejidos, en particular el oído interno y la glándula tiroides. En la tiroides, se cree que la pendrina transporta iones de yoduro fuera de sus células. El yoduro es necesario para la producción normal de hormonas tiroideas.37 En aproximadamente el 30% de los pacientes, la dishormonogénesis está presente al nacer y se diagnostica mediante un cribado neonatal de HC. Puede ser causada por mutaciones homocigotas o heterocigotas en el gen SLC26A4.38 Aún no se ha observado una clara correlación genotipo-fenotipo; se ha propuesto que el deterioro de la función tiroidea se debe a la reducción de la organificación del yodo, lo que conduce al desarrollo de una glándula agrandada, con o sin producción alterada de hormona tiroidea.
SLC5A5
El gen SLC5A5 proporciona instrucciones para producir una proteína llamada simportador de sodio y yoduro (NIS, Na + /I - symporter).39 Esta proteína transporta yoduro, una especie química del yodo cargada negativamente, a las células de ciertos tejidos. La proteína NIS se encuentra principalmente en la glándula tiroides; está involucrada en asegurar que el yodo de la dieta se acumule en la glándula tiroides para la producción de hormonas tiroideas. Este sistema depende de que la proteína NIS se coloque en la membrana celular, por lo que puede transportar yoduro desde el torrente sanguíneo a determinadas células tiroideas llamadas células foliculares. Los cambios en la expresión o función de NIS, debido a mutaciones en el gen SLC5A5, causan un amplio espectro de trastornos tiroideos.3 Los criterios de diagnóstico sugeridos para el defecto de transporte de yoduro incluyen: bocio pequeño o grande con hipotiroidismo o hipotiroidismo compensado, captación tiroidea reducida o ausente de radioyodo, incapacidad para concentrar yoduro en las glándulas salivales y respuesta positiva a terapia con altas dosis de yoduro.40 Hasta la actualidad se han comunicado 15 mutaciones genéticas inactivadoras del gen SLC5A5.41,42
La nueva era de la secuenciación de próxima generación (NGS, next-generation sequencing) ha permitido que algunas investigaciones analizaran la importancia de estos genes de manera global; por ejemplo, un estudio demostró que alrededor del 96.8% de los pacientes con HC tenían, al menos, una variante potencialmente patogénica para los genes relacionados con dishormonogénesis (DUOX2, TG, TPO y SLC26A4), mientras que el porcentaje de variantes patogénicas fue ampliamente menor en genes relacionados con disgenesia tiroidea.43 Esto contradice a un estudio anterior en el cual se encontró que entre el 80% y el 85% de la población con HC presentaba mutaciones en genes de disgenesia tiroidea, como TSHR, PAX8, NKX2-1, NKX2-5 y FOXE1.44 La discrepancia puede reflejar simplemente el hecho de que no se han generado datos suficientes, y también a que ciertos factores de riesgo genético difieren significativamente de un grupo étnico a otro.
Epidemiología del hipotiroidismo congénito
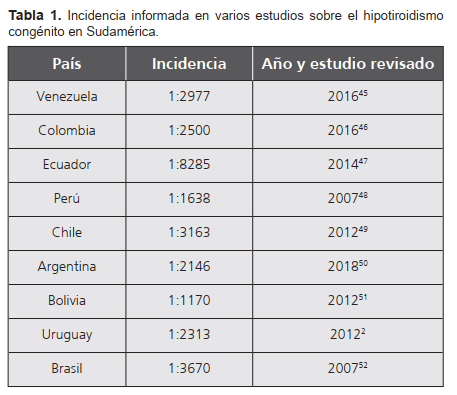
A nivel global, la prevalencia de HC se informa de 1 por cada 2000 a 1 por cada 4000 recién nacidos, con ciertas variaciones, de acuerdo con los estudios revisados, en la información disponible de los países sudamericanos, los cuales se resumen en la Tabla 1.
Diagnóstico clínico
El HC es una endocrinopatía que produce la discapacidad cognitiva prevenible más frecuente en el recién nacido.53 El pronóstico del desarrollo neurológico se relaciona en forma inversa con la edad de diagnóstico e inicio de tratamiento de la enfermedad. La explicación de que los síntomas del HC sean de apariencia poco pronunciada al nacer, inclusive en casos graves de hipotiroidismo bioquímico, consiste en el hecho de que las hormonas tiroideas maternas atraviesan la placenta durante el desarrollo neurológico del feto, de manera que lo protege de las manifestaciones iniciales significativas.53 Entre los antecedentes patológicos durante el embarazo, hasta el 20% pueden presentar embarazo prolongado. El peso y la longitud al nacimiento son generalmente normales, aunque pueden tener mayor circunferencia craneal. Por esta razón, se han desarrollado programas de tamizaje neonatal en todo el mundo para la detección oportuna de esta afección.54 Sin embargo, en caso de que el HC no se investigue por medio de una detección sistemática, en los tres primeros meses de vida el recién nacido presentará signos de letargia; hipotonía; llanto ronco; piel seca, moteada e ictericia prolongada; lengua agrandada (macroglosia); hernia umbilical y estreñimiento.54 El cuello debe examinarse en busca de bocio, y su presencia sugeriría dishormonogénesis como base del HC.24
Tratamiento
Se ha demostrado que la introducción temprana de un tratamiento para el HC con LT4, dentro de las dos primeras semanas de vida, es esencial para asegurar un adecuado desarrollo neurológico y un normal coeficiente intelectual. La introducción del tratamiento tardío puede ir asociado con alteración del desarrollo psiconeuromotor.55,56 El índice de biodisponibilidad de LT4 administrada por vía oral es, en una persona normal, de entre el 50% y el 80%.57 Las elevadas dosis iniciales de LT4 promueven el desarrollo psiconeuromotor adecuado para los bebés con HC. No se han registrado efectos adversos con dosis entre 10 y 15 µg/kg/día e incluso superiores. El uso de dosis iniciales inferiores a 10 µg/kg/día de LT4 en el HC puede causar deficiencias en el desarrollo.58 Es conveniente llevar a cabo una vigilancia frecuente de las dosis para asegurar que se ajusten en el tratamiento con L-tiroxina, y así evitar una infradosificación o sobredosificación que afecte negativamente las funciones neurosensoriales.58
Conclusiones
Los artículos revisados aportan información sobre los determinantes genéticos que se han asociado con la aparición de HC. Se han identificado 12 genes involucrados y se han descrito varias mutaciones y anomalías para la disgenesia tiroidea y la dishormonogénesis. La prevalencia de HC en Sudamérica varía, con la más baja informada en Ecuador y la más alta en Bolivia. El tamizaje neonatal del HC representa el método más eficaz para prevenir el retraso mental y garantizar niveles de coeficiente intelectual normales en esta población de pacientes. Su tratamiento consiste esencialmente en la administración inicial alta de LT4; sin embargo, es conveniente llevar a cabo una vigilancia frecuente de las dosis para asegurar que estas se ajusten de manera de una infradosificación o sobredosificación.

