











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkI. INTRODUCCIÓN
Las aleaciones base zirconio son utilizadas en los reactores nucleares debido a su baja absorción de neutrones y buena respuesta a la corrosión a alta temperatura,1 en particular, la aleación Zr-2,5%Nb es utilizada en la fabricación de los tubos de presión de las centrales nucleares tipo CANDU (CANadá Deuterio Uranio).2 En éstas, el hidrógeno altera la mayorı́a de las propiedades mecánicas del material y puede llevar a la rotura catastrófica del componente a través del proceso conocido como fisuración asistida por hidruros (DHC). Teniendo en cuenta que el hidrógeno puede estar presente en las aleaciones como una impureza remanente del proceso de fabricación y además puede ingresar durante la vida en servicio del material, es de suma importancia evaluar la respuesta de las aleaciones frente al hidrógeno.3 Así, a fin de evaluar el daño producido al componente y pronosticar su vida útil en servicio, resulta fundamental poder caracterizar la difusión del H en el material. En estas condiciones, los tubos son sometidos a temperatura e irradiación, que modifican sus propiedades mecánicas, microestructura, solubilidad y coeficiente de difusión del H, lo cual afecta directamente la DHC.
La aleación Zr-2.5Nb es bifásica con granos de fase Zr- rodeados de láminas de Zr-
rodeados de láminas de Zr-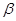 .4 La Fig. muestra micrografías del material de los tubos de presión, obtenidas por microscopía electrónica de transmisión (TEM). Las láminas originales de Zr- son metaestables en el rango de temperaturas de funcionamiento del reactor.
.4 La Fig. muestra micrografías del material de los tubos de presión, obtenidas por microscopía electrónica de transmisión (TEM). Las láminas originales de Zr- son metaestables en el rango de temperaturas de funcionamiento del reactor.
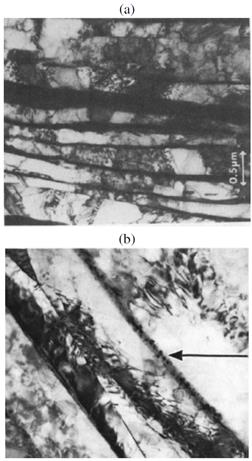
FIG. 1: Micrografías TEM del material de tubos de presión tomadas de (a) ref. 4 y (b) ref. 1. La flecha señala pequeños precipitados de Zr-ω en una matriz de Zr-β enriquecida.
Por esta razón se degradan con el tiempo perdiendo su continuidad, e interrumpiendo ası́ los caminos (supuestamente) rápidos para la difusión del hidrógeno en la dirección longitudinal del tubo, al mismo tiempo que se modifica su contenido de Nb. Los experimentos realizados en nuestro laboratorio5 plantean dos importantes interrogantes que pretendemos responder en este trabajo, ellos son:
-La pérdida de continuidad de las láminas originales ¿es efectivamente la responsable de la disminución observada en el coeficiente de difusión del H a medida que trascurre el tiempo a una dada temperatura?
-La difusión y/o solubilidad del hidrógeno en la aleación ¿se ven afectadas por el contenido de Nb de la fase Zr-
?
En las secciones que siguen presentamos las metodologías que utilizamos para abordar estas cuestiones y los resultados que de ellas se derivan. Brevemente, i) desarrollamos un modelo fenomenológico de la difusión en los tubos de presión, con el fin de evaluar la influencia de la fase continua Zr- sobre el coeficiente de difusión del hidrógeno, tanto axial como radial; y ii) empleamos la teoría del estado de transición6 (TST), provista de parámetros calculados ab initio mediante el código SIESTA,7 para estudiar el coeficiente de difusión en el Zr- conforme varía la concentración de Nb.
II. METODOLOGÍA
Modelo de la difusión en tubos
Proponemos un modelo de difusión unidimensional, Fig. 2, basado en el de Skinner y Dutton (S&D),1 que consiste de un arreglo tridimensional formado por un paralelepípedo de fase Zr-, rodeado en tres de sus caras por finas láminas,  y
y  , de una fase bifásica, que llamaremos
, de una fase bifásica, que llamaremos 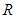 a secas, compuesta por 31% de Zr-
a secas, compuesta por 31% de Zr-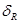 enriquecida y 69% de Zr-
enriquecida y 69% de Zr-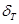 según los resultados de S&D.1 Esto último correspondería a la condición del tubo luego del tratamiento ordinario en autoclave a 400 ºC por 24 h, previo al ingreso en reactor (c.f. Fig. 1). Dichas láminas cumplen la función de caminos rápidos para la difusión del H, en consonancia con el modelo original de Sawatsky et al.4
según los resultados de S&D.1 Esto último correspondería a la condición del tubo luego del tratamiento ordinario en autoclave a 400 ºC por 24 h, previo al ingreso en reactor (c.f. Fig. 1). Dichas láminas cumplen la función de caminos rápidos para la difusión del H, en consonancia con el modelo original de Sawatsky et al.4
Observamos que en cualquier dirección del tubo, el camino efectivo de difusión es una sucesión de granos 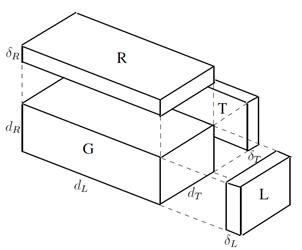 seguidos de finas láminas
seguidos de finas láminas 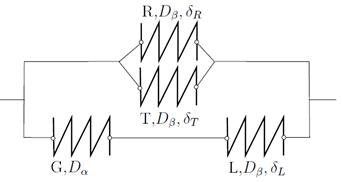 , en paralelo con un camino continuo compuesto por láminas
, en paralelo con un camino continuo compuesto por láminas 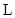 únicamente. Por ejemplo, en la dirección longitudinal el grano
únicamente. Por ejemplo, en la dirección longitudinal el grano 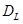 está en serie con la lámina
está en serie con la lámina  de espesor
de espesor  , y a su vez este conjunto está en paralelo con las láminas
, y a su vez este conjunto está en paralelo con las láminas  y
y 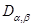 de espesores
de espesores  y
y 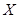 respectivamente. La Fig. 3 muestra el análogo eléctrico del caso.
respectivamente. La Fig. 3 muestra el análogo eléctrico del caso.

Considerando que el coeficiente de difusión desempeña el papel de una conductividad, la resolución del circuito conduce a la siguiente expresión del coeficiente de difusión efectivo en la dirección longitudinal del tubo, 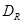 ,
,
(1)
donde  y
y
(2)
En las Ecs. (1) y (2),  son coeficientes de difusión,
son coeficientes de difusión,  es la fracción volumétrica de lámina
es la fracción volumétrica de lámina  ,
,  es el coeficiente de partición de solubilidades del H en fase
es el coeficiente de partición de solubilidades del H en fase 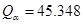 respecto de
respecto de  . Una expresión análoga se obtiene para
. Una expresión análoga se obtiene para 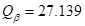 sin más que intercambiar
sin más que intercambiar  con
con 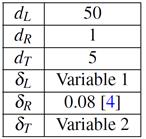 .
.
Resumimos a continuación los datos del modelo. Para las difusividades ,  , (
, (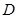 m
m /s y
/s y 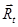 KJ/mol):
KJ/mol): 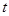 ,
,  de ref.8 y
de ref.8 y 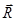 ,
,  de ref..1 Para el coeficiente de partición usamos
de ref..1 Para el coeficiente de partición usamos  .4 Los datos geométricos se consignan en la Tabla 1 (c.f. Fig. 2).
.4 Los datos geométricos se consignan en la Tabla 1 (c.f. Fig. 2).
Las dimensiones del grano 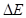 respetan proporciones típicas para los tubos.9 Las variables 1 y 2 se fijan buscando el mejor ajuste con las mediciones de cociente de velocidades de propagación de fisuras (
respetan proporciones típicas para los tubos.9 Las variables 1 y 2 se fijan buscando el mejor ajuste con las mediciones de cociente de velocidades de propagación de fisuras ( ) en sentido longitudinal vs. radial; según Dutton et al.10
) en sentido longitudinal vs. radial; según Dutton et al.10
 es proporcional a las difusividades. Además imponemos la condición que el volumen de las láminas rodeando al grano
es proporcional a las difusividades. Además imponemos la condición que el volumen de las láminas rodeando al grano 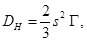 no debe exceder el 10% del total.11
no debe exceder el 10% del total.11
Coeficientes de difusión ab initio
Para el caso escalar, adecuado a la presente situación de redes cúbicas, el coeficiente de difusión  adopta la expresión
adopta la expresión
(3)
donde 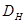 es la posición del trazador a tiempo
es la posición del trazador a tiempo  y
y 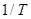 indica promedio sobre el ensamble de trayectorias. Considerando al H como un caminante aleatorio por la red BCC,
indica promedio sobre el ensamble de trayectorias. Considerando al H como un caminante aleatorio por la red BCC, 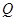 se compone de saltos elementales entre sitios tetraédricos (T) primeros vecinos, conectados por barreras de energía. En el marco de la TST6 la frecuencia
se compone de saltos elementales entre sitios tetraédricos (T) primeros vecinos, conectados por barreras de energía. En el marco de la TST6 la frecuencia  de estos saltos se expresa según,
de estos saltos se expresa según,
(4)
donde 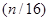 es la barrera de energía determinada por la configuración de ensilladura entre sitios T y
es la barrera de energía determinada por la configuración de ensilladura entre sitios T y  y
y  son las frecuencias de vibración del H en T (equilibrio) y en el punto silla, respectivamente. Obsérvese que para la silla se suprime la frecuencia imaginaria asociada y además hemos supuesto, dado la diferencia de masas, que el H vibra desacoplado de la red. A altas temperaturas el factor pre-exponencial se reduce al clásico cociente de frecuencias.
son las frecuencias de vibración del H en T (equilibrio) y en el punto silla, respectivamente. Obsérvese que para la silla se suprime la frecuencia imaginaria asociada y además hemos supuesto, dado la diferencia de masas, que el H vibra desacoplado de la red. A altas temperaturas el factor pre-exponencial se reduce al clásico cociente de frecuencias.
En este contexto, para el caso de una matriz BCC pura (e.g. H en Nb) la Ec. (3) toma la forma explícita,
(5)
siendo 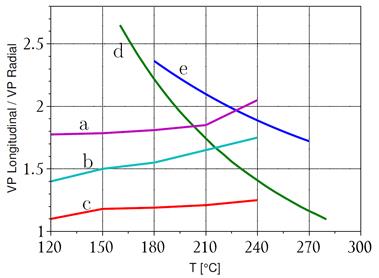 la longitud del salto. Lamentablemente las expresiones analíticas resultan imposibles ni bien la simetría se degrada, máxime en aleaciones desordenadas como el Zr-
la longitud del salto. Lamentablemente las expresiones analíticas resultan imposibles ni bien la simetría se degrada, máxime en aleaciones desordenadas como el Zr- . En este trabajo enfocamos el estudio sobre nueve aleaciones con distintos contenidos de Nb. Cada una de estas, si bien ordenada, posee un conjunto de saltos no equivalentes, razón por la cual atacamos el cálculo de
. En este trabajo enfocamos el estudio sobre nueve aleaciones con distintos contenidos de Nb. Cada una de estas, si bien ordenada, posee un conjunto de saltos no equivalentes, razón por la cual atacamos el cálculo de  mediante simulación numérica de la Ec.(3), empleando la técnica de Monte Carlo cinético12 (KMC), alimentada con frecuencias del tipo Ec.(4) en el límite clásico (suficiente para nuestros propósitos). Esto produce gráficos de Arrhenius,
mediante simulación numérica de la Ec.(3), empleando la técnica de Monte Carlo cinético12 (KMC), alimentada con frecuencias del tipo Ec.(4) en el límite clásico (suficiente para nuestros propósitos). Esto produce gráficos de Arrhenius,  vs.
vs. 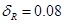 , ligeramente curvos, que ajustamos con una sola energía de activación
, ligeramente curvos, que ajustamos con una sola energía de activación 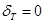 (tomada como representativa) en un intervalo de temperaturas que comprende las de operación del reactor.
(tomada como representativa) en un intervalo de temperaturas que comprende las de operación del reactor.
Calculamos todas las frecuencias y energías necesarias mediante el código SIESTA,7 basado en la teoría del funcional densidad (DFT), pseudopotenciales y bases numéricas localizadas. Los dos últimos aspectos fueron desarrollados y optimizados en este trabajo, a fin de lograr una pareja de especies Zr-Nb lo más consistente posible. La mayoría de los cálculos se realizaron sobre (super) celdas cúbicas BCC de 16 nodos ( celdillas unitarias), con distinto contenido de Nb: 0, 1, 2, 4, 8, 12, 14, 15 y 16 átomos, nueve en total, que en lo sucesivo notaremos como
celdillas unitarias), con distinto contenido de Nb: 0, 1, 2, 4, 8, 12, 14, 15 y 16 átomos, nueve en total, que en lo sucesivo notaremos como 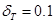 . Estas celdas fueron generadas con la máxima simetría posible a fin de minimizar el número de saltos a considerar. Para c/u de ellas encontramos el parámetro de red óptimo, luego lo fijamos e introdujimos 1 átomo de H en distintas posiciones, que a su vez optimizamos mediante relajación de coordenadas. Así determinamos que los sitios tetraédricos son mínimos de la energía, en tanto que los octaédricos (de mayor energía) resultan sillas de rango 2. En cuanto a las sillas entre dos sitios T, la mayoría puede evaluarse ubicando al H a mitad de camino sobre el segmento entre ambos y minimizando la energía con la restricción de mantener al H sobre el plano perpendicular al segmento. Para los pocos casos no simétricos recurrimos al método de “drag”,13 que consiste en levantar el perfil de energía para posiciones iniciales del H interpoladas entre dos configuraciones T relajadas, y minimizar la energía anulando la componente de la fuerza sobre el (hiper)segmento de interpolación.
. Estas celdas fueron generadas con la máxima simetría posible a fin de minimizar el número de saltos a considerar. Para c/u de ellas encontramos el parámetro de red óptimo, luego lo fijamos e introdujimos 1 átomo de H en distintas posiciones, que a su vez optimizamos mediante relajación de coordenadas. Así determinamos que los sitios tetraédricos son mínimos de la energía, en tanto que los octaédricos (de mayor energía) resultan sillas de rango 2. En cuanto a las sillas entre dos sitios T, la mayoría puede evaluarse ubicando al H a mitad de camino sobre el segmento entre ambos y minimizando la energía con la restricción de mantener al H sobre el plano perpendicular al segmento. Para los pocos casos no simétricos recurrimos al método de “drag”,13 que consiste en levantar el perfil de energía para posiciones iniciales del H interpoladas entre dos configuraciones T relajadas, y minimizar la energía anulando la componente de la fuerza sobre el (hiper)segmento de interpolación.
En todas estas simulaciones con SIESTA empleamos los siguientes parámetros principales: i) paso de malla espacial equivalente a 450 Ry (MeshCutoff), ii) temperatura electrónica de 0.15 eV (distribución de Fermi-Dirac), iii) malla en espacio recíproco de  puntos, iv) fuerzas residuales por debajo de 0.01 eV/Å para relajaciones convergidas.
puntos, iv) fuerzas residuales por debajo de 0.01 eV/Å para relajaciones convergidas.
III. RESULTADOS Y DISCUSIÓN
Modelo fenomenológico
La Fig. 4 muestras difusividades axiales del hidrógeno para tubos bajo distintas condiciones, ya sea medidas, en líneas llenas, o predichas por modelos, en líneas de trazos. La curva (a) corresponde a Zr- ,8 (b) y (c) corresponden a Zr-2.5wt%Nb con tratamientos térmicos de 565 ºC y 400 ºC respectivamente,1 (d) es la medición de 4 también para Zr-2.5wt%Nb, (e) es la predicción de,4 (f) la de1 y (g) la del presente trabajo. Para esta última, las variables 1 y 2 de la Tabla 1 se fijaron en 0.08, lo que resulta en una fracción de volumen de 9% para la fase
,8 (b) y (c) corresponden a Zr-2.5wt%Nb con tratamientos térmicos de 565 ºC y 400 ºC respectivamente,1 (d) es la medición de 4 también para Zr-2.5wt%Nb, (e) es la predicción de,4 (f) la de1 y (g) la del presente trabajo. Para esta última, las variables 1 y 2 de la Tabla 1 se fijaron en 0.08, lo que resulta en una fracción de volumen de 9% para la fase  , por debajo del máximo permitido de 10%11. El rango de temperaturas de 453 a 543 K es el que en general se utiliza para los ensayos de DHC.11
, por debajo del máximo permitido de 10%11. El rango de temperaturas de 453 a 543 K es el que en general se utiliza para los ensayos de DHC.11
FIG. 4:
 axial medido para tubos en distintas condiciones, líneas llenas, y predicciones de modelos, líneas de trazos. Detalles en texto principal.
axial medido para tubos en distintas condiciones, líneas llenas, y predicciones de modelos, líneas de trazos. Detalles en texto principal.
Observamos que nuestro modelo concuerda muy bien con los resultados experimentales de Sawatsky et al.4
Por otra parte, en la Fig. 5 mostramos el cociente de 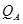 axial/radial medido por Jovanović et al.,14 curvas (a), (b) y (c), en material Zr-2.5wt%Nb de tubos con tratamientos térmicos a 400 ºC por 1 h, 24 h y 1000 h, respectivamente. La curva (d) es lo mismo pero según la norma canadiense CSA 285.15;15 finalmente (e) es nuestra predicción para el cociente de difusividades axial/radial. Esta última tiene un comportamiento parecido al de la norma canadiense para el cociente de
axial/radial medido por Jovanović et al.,14 curvas (a), (b) y (c), en material Zr-2.5wt%Nb de tubos con tratamientos térmicos a 400 ºC por 1 h, 24 h y 1000 h, respectivamente. La curva (d) es lo mismo pero según la norma canadiense CSA 285.15;15 finalmente (e) es nuestra predicción para el cociente de difusividades axial/radial. Esta última tiene un comportamiento parecido al de la norma canadiense para el cociente de 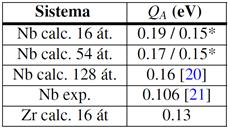 y está dentro de los valores consignados en literatura para esta magnitud.1,8 Ambas difieren sin embargo de los resultados de Jovanović et al., curvas (a) y (b), aunque también dentro del rango de valores. Al presente, las razones de esta discrepancia son de carácter sumamente especulativo.
y está dentro de los valores consignados en literatura para esta magnitud.1,8 Ambas difieren sin embargo de los resultados de Jovanović et al., curvas (a) y (b), aunque también dentro del rango de valores. Al presente, las razones de esta discrepancia son de carácter sumamente especulativo.
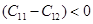
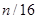
Respetando la limitación mencionada respecto de la fracción volumétrica de las fases presentes, es posible ajustar los espesores  ,
,  , y
, y 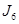 de nuestro modelo con el fin de alcanzar otros valores máximos y mínimos de cociente axial/radial para
de nuestro modelo con el fin de alcanzar otros valores máximos y mínimos de cociente axial/radial para 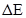 . El rango de valores se muestra de forma esquemática en la Fig. 6. Los valores máximos fueron alcanzados con
. El rango de valores se muestra de forma esquemática en la Fig. 6. Los valores máximos fueron alcanzados con 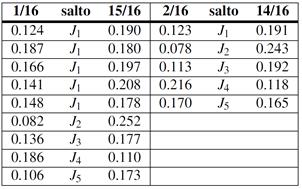 ,
,  , y
, y  , obteniendo un volumen para las láminas que rodean al grano de Zr-
, obteniendo un volumen para las láminas que rodean al grano de Zr-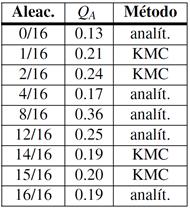 de 7.4%; los valores mínimos fueron logrados para
de 7.4%; los valores mínimos fueron logrados para  ,
,  y
y  , obteniendo una fracción volumétrica de aproximadamente 10%.
, obteniendo una fracción volumétrica de aproximadamente 10%.
Cálculos ab initio preliminares
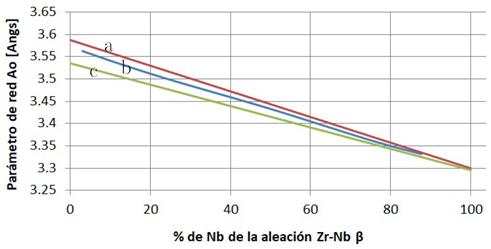
A fin de validar nuestras predicciones, en primer término realizamos cálculos de parámetros de red y energía de activación para 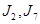 en Nb puro, dado la existencia de resultados experimentales confiables así como de cálculos de literatura. En la Fig. 7 mostramos la variación del parámetro de red en función del contenido de Nb, predicha a partir de las nueve aleaciones mencionadas, comparada con otras estimaciones ab initio16 mediante el código Wien2k,17 y una interpolación de resultados experimentales.18,19 Nuestros cálculos emplearon celdas cúbicas de 16 nodos, los de ref.16 celdas no cúbicas de 32 nodos, y los experimentos corresponden a aleaciones desordenadas retenidas desde temperaturas y concentraciones de Nb que estabilizan la fase BCC (
en Nb puro, dado la existencia de resultados experimentales confiables así como de cálculos de literatura. En la Fig. 7 mostramos la variación del parámetro de red en función del contenido de Nb, predicha a partir de las nueve aleaciones mencionadas, comparada con otras estimaciones ab initio16 mediante el código Wien2k,17 y una interpolación de resultados experimentales.18,19 Nuestros cálculos emplearon celdas cúbicas de 16 nodos, los de ref.16 celdas no cúbicas de 32 nodos, y los experimentos corresponden a aleaciones desordenadas retenidas desde temperaturas y concentraciones de Nb que estabilizan la fase BCC ( K y
K y  resp.). Ambos cálculos muestran un razonable acuerdo con los experimentos, reproduciendo en particular el comportamiento lineal tipo ley de Vegard, si bien el nuestro con una subestimación un poco mayor.
resp.). Ambos cálculos muestran un razonable acuerdo con los experimentos, reproduciendo en particular el comportamiento lineal tipo ley de Vegard, si bien el nuestro con una subestimación un poco mayor.
FIG. 7: Parámetro de red vs. contenido de Nb; (a) ajuste a datos experimentales,18,19 (b) cálculo de ref.,16 (c) cálculo presente.
En la Tabla 2 presentamos energías de activación  para las especies puras, calculadas y medida en Nb. Nuestros valores marcados con (*) representan una extrapolación a celda de tamaño infinito, basada en teoría elástica y siguiendo metodologías de literatura.22,23 Su coincidencia para los dos tamaños que probamos, 16 y 54 átomos, indica que debe considerarse como el (mejor) valor predicho por nuestra técnica. Observamos además un muy buen acuerdo con valores de otros autores20 utilizando celdas más grandes y el código VASP24. Ambas estimaciones, sin embargo, difieren significativamente de los (abundantes) resultados experimentales.21 Este hecho sugiere que los cálculos de
para las especies puras, calculadas y medida en Nb. Nuestros valores marcados con (*) representan una extrapolación a celda de tamaño infinito, basada en teoría elástica y siguiendo metodologías de literatura.22,23 Su coincidencia para los dos tamaños que probamos, 16 y 54 átomos, indica que debe considerarse como el (mejor) valor predicho por nuestra técnica. Observamos además un muy buen acuerdo con valores de otros autores20 utilizando celdas más grandes y el código VASP24. Ambas estimaciones, sin embargo, difieren significativamente de los (abundantes) resultados experimentales.21 Este hecho sugiere que los cálculos de 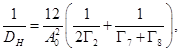 aquí presentados difícilmente puedan ser cuantitativamente comparados contra experimentos. Las sutilezas que rodean los aspectos cuantitativos de este tipo de cálculos para el H, ya fueron observadas por nosotros25 en el sistema Fe-Cr, de similar estructura cristalina que el presente.
aquí presentados difícilmente puedan ser cuantitativamente comparados contra experimentos. Las sutilezas que rodean los aspectos cuantitativos de este tipo de cálculos para el H, ya fueron observadas por nosotros25 en el sistema Fe-Cr, de similar estructura cristalina que el presente.
Por otra parte, el cálculo de 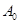 para Zr BCC también presenta problemas. Esta fase es elásticamente inestable a 0 K,
para Zr BCC también presenta problemas. Esta fase es elásticamente inestable a 0 K, 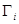 . Ambos códigos, SIESTA y VASP (resultados propios), arrojan sin embargo valores positivos muy pequeños de este módulo. Entendemos es por esta razón que, al introducir el H en el sitio T de celdas mayores que 16 átomos, las mismas adquieren grandes deformaciones espurias. Resulta claro además, que las extrapolaciones elásticas en estas circunstancias serían de validez dudosa.
. Ambos códigos, SIESTA y VASP (resultados propios), arrojan sin embargo valores positivos muy pequeños de este módulo. Entendemos es por esta razón que, al introducir el H en el sitio T de celdas mayores que 16 átomos, las mismas adquieren grandes deformaciones espurias. Resulta claro además, que las extrapolaciones elásticas en estas circunstancias serían de validez dudosa.
Resumiendo, en virtud de los dos párrafos previos decidimos, para todos los cálculos de barreras de energía que siguen, fijar el tamaño de celda en 16 átomos y no aplicar correcciones elásticas. De este modo trabajamos con un conjunto de valores comparables entre sí, con el cual pretendemos al menos reproducir correctamente comportamientos cualitativos.
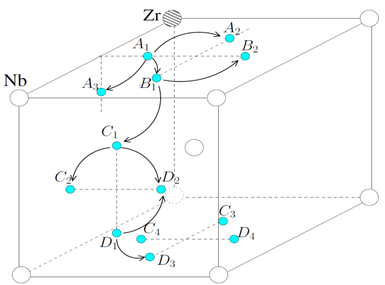
Aleaciones ordenadas: barreras de energía
Geométricamente podemos identificar las aleaciones estudiadas, 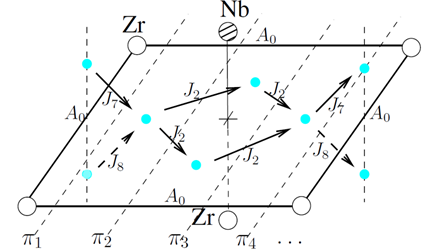 , mediante las coordenadas de la especie minoritaria dentro de la supercelda de
, mediante las coordenadas de la especie minoritaria dentro de la supercelda de 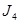 . En unidades de parámetro de red,
. En unidades de parámetro de red,
1/16 y 15/16: (0,0,0) 2/16 y 14/16: (0,0,0) (1,1,1) 4/16 y 12/16: (0.5,0.5,0.5) (1.5,1.5,0.5) (1.5,0.5,1.5) (0.5,1.5,1.5) 8/16: como en estructura B2.
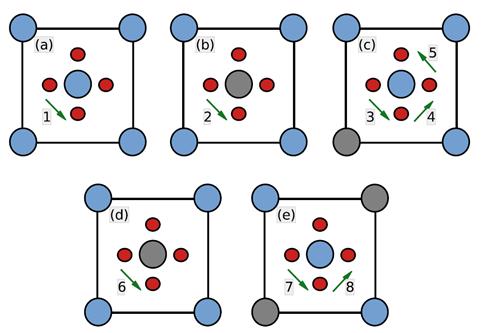
A fin de revelar posibles sistemáticas y determinar la posibilidad de su aplicación a aleaciones aleatorias, caracterizamos las barreras atendiendo a los octaedros (entorno cercano) centrados en sitios O, dentro de los cuales ocurren los saltos. Para las aleaciones consideradas existen 5 de estos octaedros, que comprenden un total de 8 saltos, identificados como  y representados esquemáticamente en la Fig. 8.
y representados esquemáticamente en la Fig. 8.
Las figuras muestran una proyección en planta del sitio O de la red BCC. Los círculos pequeños indican sitios T, sobre una cara de la celdilla unitaria corriente. Los círculos más grandes representan átomos de Nb o Zr, aquellos en el centro quedan por arriba y por debajo del plano del papel; en (b) esos 2 átomos son distintos y en (d) iguales. Finalmente, la especie minoritaria aparece en gris.
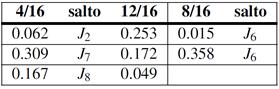
Las Tablas 3 y 4 reúnen todos nuestros cálculos de barreras. La barrera asociada a un dado salto 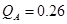 no es necesariamente simétrica respecto del sentido del salto. A los fines de estas tablas tomamos el valor medio de ambos sentidos. Para el cálculo del coeficiente de difusión en cambio (e.g. via KMC), tales asimetrías son consideradas. En todos los casos en que un dado
no es necesariamente simétrica respecto del sentido del salto. A los fines de estas tablas tomamos el valor medio de ambos sentidos. Para el cálculo del coeficiente de difusión en cambio (e.g. via KMC), tales asimetrías son consideradas. En todos los casos en que un dado  se repite, los saltos respectivos son equivalentes desde el punto de vista del octaedro, siempre y cuando no se cruce la composición del 50%; en este último caso los octaedros asociados al salto
se repite, los saltos respectivos son equivalentes desde el punto de vista del octaedro, siempre y cuando no se cruce la composición del 50%; en este último caso los octaedros asociados al salto 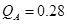 resultan complementarios (Fig. 8(d)).
resultan complementarios (Fig. 8(d)).
De la inspección de las tablas surge claro un comportamiento sistemático de las energías de salto, como así también que el entorno cercano definido por el octaedro resulta algo deficiente para la predicción cuantitativa. En general las aleaciones ricas en Nb muestran un comportamiento más consistente a lo largo de las distintas concentraciones, siendo más notable la dispersión de valores para las ricas en Zr. Nótese en particular lo que sucede con , donde las para las aleaciones ricas en Nb prácticamente coinciden con el valor de Nb puro, Tabla 2, no siendo así para las ricas en Zr. Queda para la especulación si este resultado está o no relacionado con la (casi) inestabilidad elástica del Zr BCC mencionada anteriormente.
Aleaciones ordenadas: coeficientes de difusión
Con las barreras disponibles nos abocamos al cálculo de los coeficientes de difusión, , en particular determinamos una energía de activación efectiva, , por ajuste de Arrhenius en el intervalo entre 300 K y 1000 K. Los resultados de este procedimiento se consignan en la Tabla 5. Para algunas aleaciones el cálculo puede hacerse analíticamente, en otros casos utilizamos KMC, condición que también se indica en la tabla. A modo de ejemplo, a continuación detallamos un caso de KMC y otro analítico de cierto interés.
Para la composición 15/16 se identifican 4 sitios T no equivalentes, , en la Fig. 9, de acuerdo a su posición respecto de un átomo de Zr. La figura también indica todos los saltos no equivalentes, en el sentido en que ingresan al código KMC. Nótese que sitios equivalentes pueden estar conectados por saltos no equivalentes. Por otra parte, la frecuencia del salto inverso queda determinada por la condición de balance detallado, dado las energías relativas de los sitios T involucrados. Para este caso particular, los parámetros más relevantes del cálculo fueron,
-Frecuencia de ataque:
Hz-Matriz de barreras (eV):
-
(6)
-Número de trayectorias: 10000
-Número de saltos/trayectoria: 10000
Los 2 últimos aseguran un error en inferior a 1%.
La composición 4/16 admite resolución analítica. La estructura cristalina consiste de celdillas BCC unitarias conteniendo Nb en su centro, rodeadas en sus 6 caras por celdillas conteniendo Zr (mayoritario) y viceversa. Esa simetría produce que los sitios T sean todos equivalentes, y los saltos entre ellos simétricos: y de Tabla 4. De este modo identificamos caminos de migración independientes, paralelos a los ejes Cartesianos, que pueden interpretarse como una sucesión de un paralelo de saltos en serie con un paralelo entre y , representados en la Fig. 10. Luego, aplicando ideas similares a las del modelo fenomenológico, obtenemos una expresión cerrada para ,
(7)
donde es el parámetro de red de la aleación y las frecuencias de salto. Incidentalmente, para esta composición resulta un cortocircuito relativo, y siendo la barrera de muy alta, queda dominado por . De ahí el pozo en respecto de las concentraciones vecinas, c.f. Tabla 5. Obsérvese que para la aleación espejo en cambio domina .
Los gráficos de Arrhenius vs. presentan una leve desviación respecto del comportamiento lineal, no obstante es posible identificar los saltos cuyas barreras dominan . En la Fig. 11 reunimos todas las barreras y energías de activación calculadas. Aparte de los casos y recién mencionados, observamos que es dominante para las composiciones de Nb y Zr puros (obviamente), 14/16 y 15/16; domina en las aleaciones 1/16 y 2/16; finalmente domina para 50%.
Por último ya mencionamos que no esperamos acuerdo cuantitativo de nuestros cálculos con los escasos valores experimentales, e.g. eV para Zr-,4 o eV para la fase bifásica de 31% Zr- enriquecido +69% Zr-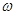 .1 No obstante, en la Fig. mostramos nuestros resultados para vs. % de Nb para diversas temperaturas. Este gráfico resulta similar a otro calculado por Samin et al.26 en el sistema Fe-Cr, BCC desordenado, utilizando otras metodologías (mucho más costosas). En literatura se ha notado1 que a medida que la fase se enriquece en Nb, disminuye el volumen de la misma y eventualmente pierde continuidad, lo cual produce un descenso de . La Fig. 12 sugiere que dicho descenso también ocurriría de modo intrínseco, por lo menos hasta alcanzar 50% de Nb.
.1 No obstante, en la Fig. mostramos nuestros resultados para vs. % de Nb para diversas temperaturas. Este gráfico resulta similar a otro calculado por Samin et al.26 en el sistema Fe-Cr, BCC desordenado, utilizando otras metodologías (mucho más costosas). En literatura se ha notado1 que a medida que la fase se enriquece en Nb, disminuye el volumen de la misma y eventualmente pierde continuidad, lo cual produce un descenso de . La Fig. 12 sugiere que dicho descenso también ocurriría de modo intrínseco, por lo menos hasta alcanzar 50% de Nb.
IV. CONCLUSIONES
Brevemente, desarrollamos un modelo de difusión fenomenológico adaptado de otros existentes en la literatura y calculamos el coeficiente de difusión del H en aleaciones de Zr-Nb para diferentes concentraciones de Nb. Respecto de la primera pregunta que planteáramos al inicio en I, podemos decir que los resultados de nuestro modelo de difusión son consistentes con la hipótesis de que la disminución observada en la difusividad del H a medida que transcurre el tiempo se debe a la pérdida de continuidad de las láminas de fase que originalmente rodean a los granos del tubo de presión.
El modelo también arroja valores de la relación axial/radial para la velocidad de propagación de fisuras, comparables a valores de literatura, dentro de un marco teórico mayormente aceptado, que la relacionan con la difusividad. Esto sería una consecuencia de la textura del tubo heredada del proceso de fabricación.
Respecto de la segunda pregunta, nuestros cálculos ab initio predicen que efectivamente la difusividad del H se ve afectada por el contenido de Nb en la fase , y que varía de manera no monótona pasando por un mínimo para la concentración de 50%Nb. Esto podría convertirse en una razón adicional de la disminución de la difusividad antes mencionada. Identificamos aquí dos caminos posibles para profundizar el estudio de la cuestión: i) extender los cálculos a aleaciones realmente desordenadas y ii) estimar el efecto de los bordes de grano en términos de tamaño y temperatura a la manera de.27

