











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El Complejo Esclerosis Tuberosa (CET), se conoce también como epiloia o síndrome de Bourneville, fue mencionada por primera vez por Friedrich Daniel Von Recklinghausen en el año de 1860, pero fue Bourneville quien describió y publicó datos por lo que también es conocida por su nombre.1
Es una enfermedad neurocutánea rara perteneciente al grupo de las genodermatosis. Se hereda de manera autosómica dominante, si bien la afectación es multisistémica, los órganos más comprometidos son piel, ojos, riñones, cerebro y corazón. Causado por la mutación de dos genes TSC1 que produce tuberina y TSC2 hamartina, siendo más frecuente y más grave la afección de TSC2.2
Se caracteriza por ser un proceso hamartomatoso progresivo que origina alteraciones en la proliferación, migración, y diferenciación celular, caracterizadas por afecciones en el cito esqueleto, procesos de crecimiento y migración celular que afectan a cualquier órgano.3
CASO CLÍNICO
Se presenta un paciente masculino de 43 años de edad con antecedentes personales de retraso madurativo y epilepsia, en tratamiento con fenobarbital y clonazepam desde los cinco años de edad, en seguimiento por neurología. Refiere antecedentes patológicos familiares de hermanos con síndrome convulsivo.
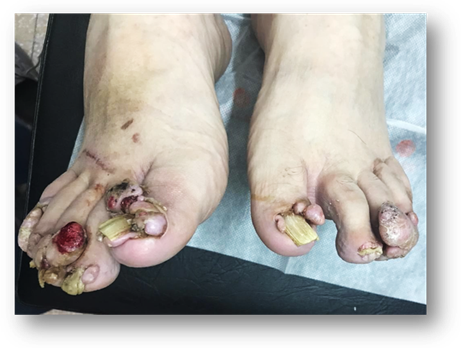
Consulta por presentar dermatosis de años de evolución caracterizada por tumoraciones en ambos pies, de gran tamaño, color rosado, consistencia blanda, impetiginizadas que causan dolor y dificultad en la marcha. (Fig. 1)
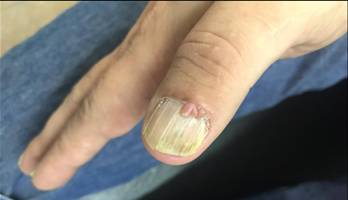
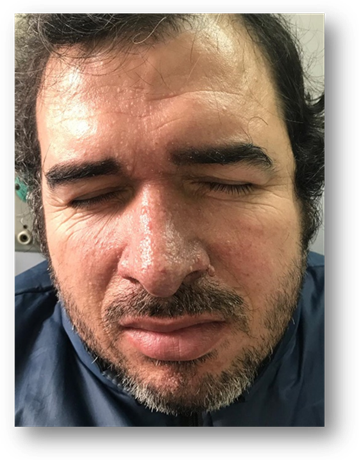
Prosiguiendo con el examen físico se evidencia en manos, sobre el lecho ungueal lesiones de las mismas características, pero de menor tamaño. (Fig. 2) En rostro sobre región centro facial se aprecian pápulas de superficiales lisas de consistencia blanda. (Fig. 3).
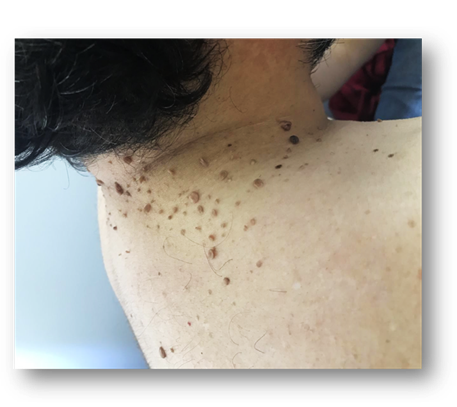
En región posterior del cuello presenta múltiples tumoraciones de diferentes tamaños, color piel, algunas hiperpigmentadas de consistencia blanda, asintomáticas. (Fig. 4).
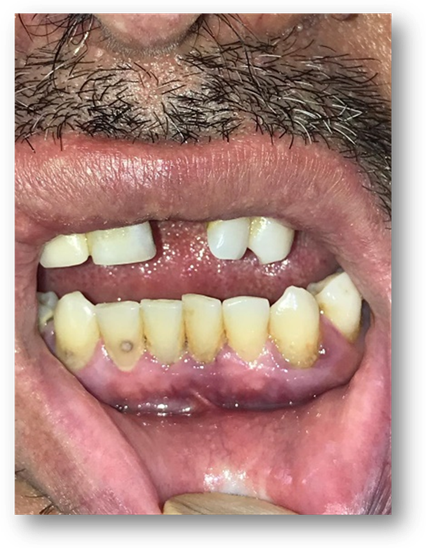
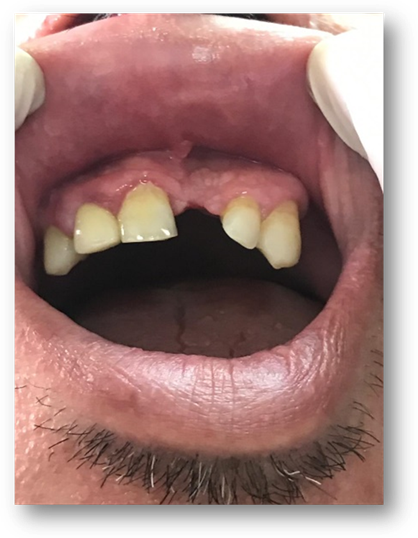
En región lumbosacra observamos una placa irregular conformada por pápulas y nódulos que coalescen tomando el aspecto de “piel de naranja”, de color piel en la periferia y amarronado en el centro. (Fig 5). Además se observan múltiples máculas hipomelanóticas en tórax y espalda, (Fig 6, 7, 8, 9), así como cavidades hipoplásicas dentarias, fibromas gingivales y máculas en papel picado (Fig 10, 11), los signos clínicos mencionados nos permiten inferir al diagnóstico de CET.
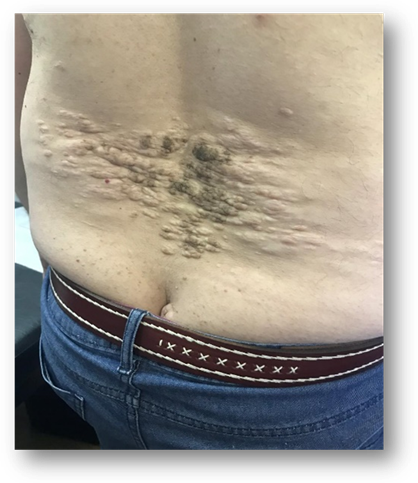
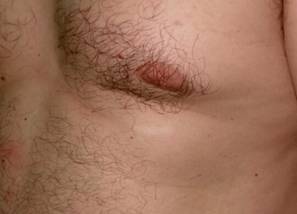
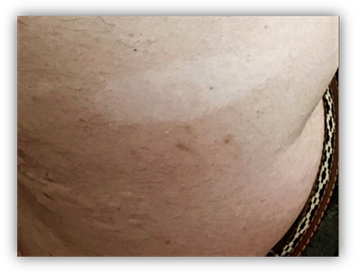
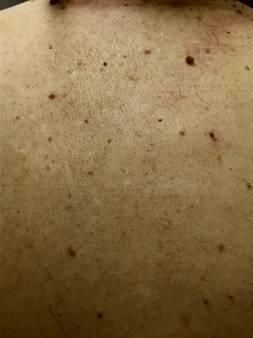
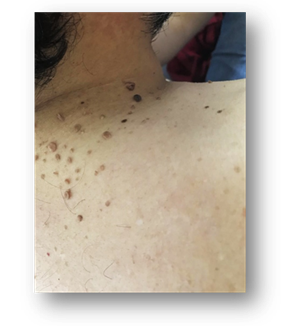
Fig 6, 7, 8 y 9: múltiples máculas hipomelanóticas en tórax y espalda.
Se realizan laboratorios de rutina con resultados normales, serologías para HIV, VDRL, hepatitis B y hepatitis C negativas.
Debido a la presencia de dolor y dificultad en la marcha se deriva a cirugía plástica en donde se realiza exéresis de las lesiones tumorales localizadas en los pies. La histopatología (Fig. 12) evidencia epidermis con hiperqueratosis y acantosis, y a mayor aumento (Fig. 13) dermis con haces de colágenos densos entrelazados que se orientan en paralelo a la epidermis.
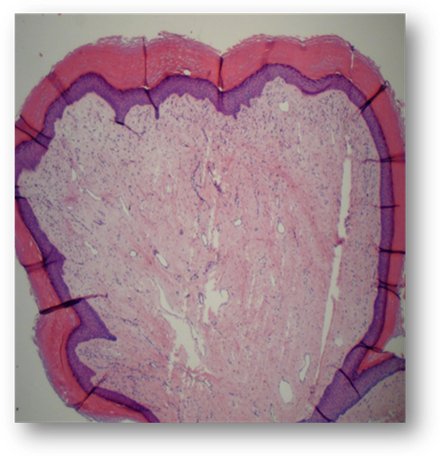
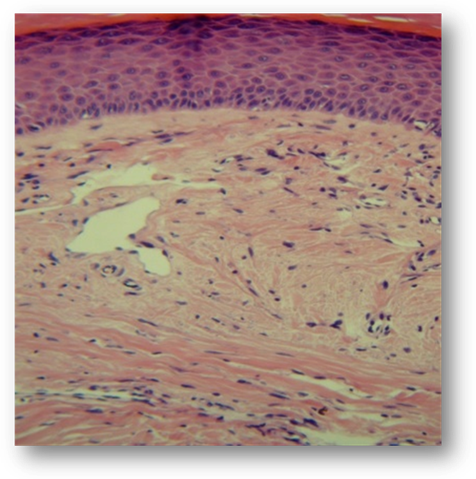
Fig 13: Histopatología: a mayor tamaño: dermis con haces de colágenos densos entrelazados que se orientan en paralelo a la epidermis.
Se decide realizar tratamiento antibiótico oral y tópico por impetiginización de lesiones tumorales, y coordinación multidisciplinaria de la atención médica, incluyendo asesoría genética.
DISCUSIÓN
El CET es una enfermedad genética autosómica dominante relativamente raro, incluido dentro de las genodermatosis, con una incidencia de uno de cada 6000 a uno de cada 10000 nacidos vivos, con una prevalencia en general de uno en 20000, e involucra a todos los grupos raciales.3
Es producido por mutaciones en un gen supresor tumoral, TSC1 ubicado en la banda cromosómica 9q34 que codifica una proteína denominada hamartina y TSC2 mapea en la banda cromosómica 16p13.3 y codifica la proteína tuberina. TSC1 y TCS2 forman un complejo que inhibe la señalización a través de la vía de la rapamicina (mTOR). La pérdida de la función de TSC1/TCS2 conduce a un aumento en la señalización de mTOR y un mayor crecimiento celular.4,5
Se caracteriza por ser un proceso hamartomatoso progresivo que origina alteraciones en la proliferación, migración y diferenciación celular caracterizadas por afecciones en citoesqueleto, procesos de crecimiento y migración celular que afectan múltiples órganos incluidos piel, cerebro, riñones, ojos, corazón y pulmones.2, 3
Presenta la triada de epilepsia, retraso madurativo y angiofibromas, siendo más frecuente la epilepsia, por tal motivo también es denominado Epiloia (epi= epilepsia, low: baja I: inteligencia, A: angiomas) descripta por Sherlok unos años más tarde.6
El diagnóstico es clínico, y consiste en relacionar la signosintomatología observadas en el paciente, con los criterios principales y secundarios evidenciados en el consenso internacional del CET del 2012 y modificados en el 2021.7 Se considera diagnóstico dos criterios principales o un criterio principal y dos secundarios, aunque también se pueden realizar con pruebas genéticas. La identificación de una variante patogénica en TSC1 o TSC2 es suficiente para el diagnóstico de la enfermedad, independientemente de los hallazgos clínicos, debido a que el diagnóstico del CET surge con el tiempo a varias edades; sin embargo, si un paciente cumple los criterios clínicos, estas no se requieren (cuadro I).7,8,9Aunque puede proporcionar información útil para otros miembros de la familia. Las pruebas genéticas de TSC1 Y TSC2 son positivas en el 75 a 95 % de los casos de las personas afectadas.3, 10
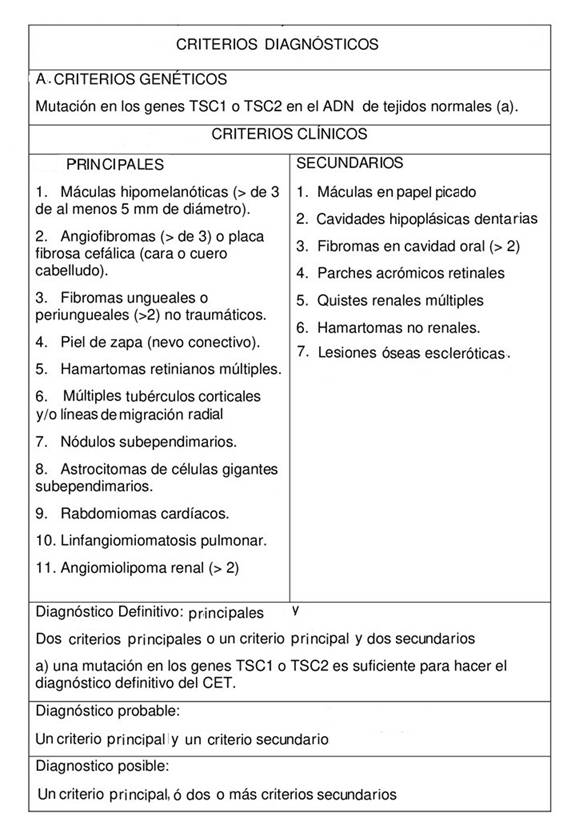
Cuadro I: Criterios diagnósticos según el consenso internacional del CET 2012, modificados en el 2021.
Los criterios diagnósticos clínicos principales incluyen: máculas hipocrómicas (tres o más, de al menos 5 mm de diámetro), angiofibromas faciales (tres o más) o placa fibrosa cefálica, fibromas ungueales (dos o más), piel de zapa o chagrín, hamartomas retinianos múltiples, múltiples tubérculos corticales y/o líneas de migración radial, (reemplaza a displasias corticales, modificación en el 2021 del consenso internacional del CET del 2012).7nódulos subependimarios, astrocitoma subependimario de células gigantes, rabdomioma cardíaco, linfangioleiomiomatosis y angiomiolipomas (2 o más).7,9
Dentro de los criterios secundarios se encuentran: lesiones cutáneas hipomelanóticas en papel picado, cavidades hipoplásicas dentarias (tres o más), fibromas en cavidad oral (dos o más), placa acrómica en retina, quistes renales múltiples, hamartoma no renal, y lesiones óseas escleróticas.7, 10
Cabe destacar que nuestro paciente presentó cuatro criterios principales (angiofibromas faciales, tumores de Köenen, tres o más máculas hipomelanóticas, piel de zapa y tres secundarios cavidades hipoplásicas dentarias, fibromas gingivales y máculas en papel picado).
El CET es una enfermedad con gran variabilidad de expresión fenotípica. No se conoce signo patognomónico de esta enfermedad, ni se suelen observar los mismos signos clínicos en los pacientes.
Las manifestaciones cutáneas son graduales, ocurren en el 81 - 91 % de los casos, y no revisten riesgo de transformación maligna.8
Las máculas hipomelanóticas son el signo cutáneo más frecuente (87 - 100 %) y de aparición temprana, son color hueso y pueden manifestarse en forma únicas (hoja de fresno o lanceolada) o múltiples (máculas en papel picado), su localización habitual es en región pre tibial y antebrazos. Cuando afectan cuero cabelludo se observan zonas de poliosis. Pueden aparecer hasta los seis años y desaparecen en la vida adulta.
Los Angiofibromas faciales o adenomas sebáceos: ocupan el segundo lugar en frecuencia (75 %), aparecen en la infancia, entre los dos y seis años de edad, como pápulas milimétricas rojizas sobre un fondo eritematoso, rosadas o hiperpigmentadas. Se distribuyen simétricamente en región centro facial y se concentran en pliegues nasolabiales.2
Los fibromas ungulares o periungulares (tumores de Köenen): aparecen en la pubertad en el 20 al 50 % de los casos. Son pápulas o nódulos rojizos o color piel normal, no vinculados con traumatismo, localizados predominantemente en pies sobre el lecho ungueal o en repliegues ungueales. Si compromete la matriz, pueden producir un surco o leuconiquia longitudinal.
Fibromas orales: inician en la pubertad, afectan principalmente la encía vestibular, los localizados fuera de la encía, se sitúan principalmente en la mucosa bucal, paladar duro y dorso de la lengua. Consiste en lesiones nodulares, de 5 mm de diámetro, color rojo amarillento, o del color de la mucosa, varían en número y en tamaño.
Nevos o hamartomas de tejido conectivo (piel de zapa): se encuentran con una frecuencia del 25 a 85% y se localizan principalmente en región lumbosacra, y con menor frecuencia en nalgas y muslos, presentes en lactantes, pero se evidencian más tarde como placas irregulares amarillo-marrón o rosadas, firmes, de 1 a 10 cm de diámetro, con superficie irregular.
Placa facial fibrosa: placa blanda o firme, lisa y sobrelevada, color piel, roja o hiperpigmentada, ubicada en cuero cabelludo y región frontal, que aparece en la pubertad.
Los síntomas neurológicos presentes en el 85% de los casos, son la principal causa de morbimortalidad, dentro de estas encontramos la epilepsia y las alteraciones neuropsiquiátricas, incluidos los problemas de aprendizaje, los trastornos del aspecto autista y el trastorno de hiperactividad con déficit de atención, comúnmente vinculdas a lesiones cerebrales incluyendo hamartomas glioneuronales (también llamados tuberomas), lesiones de la sustancia blanca y astrocitomas de células gigantes subependimarios.5, 8, 11
Los tubérculos corticales, los nódulos subependimarios y las lesiones de la sustancia blanca se encuentran comúnmente en el CET. Las manifestaciones menos comunes incluyen atrofia cerebelosa, disgenesia del cuerpo calloso, malformación de Chiari, quiste aracnoideo e infartos debidos a enfermedades vasculares oclusivos.9, 12
Las manifestaciones renales están presentes en el 60-80% de los pacientes. Encontramos los angiomiolipomas, quistes renales, y carcinoma de células renales. El angiomiolipoma es la lesión renal más frecuente en los pacientes con CET (70 %) y su incidencia aumenta con la edad, representan la segunda causa de muerte. Son tumores benignos derivados de células del endotelio. Están compuestos por tejido adiposo, músculo liso y vasos sanguíneos; son pequeños, benignos, múltiples, bilaterales y asintomáticos. Pueden complicarse con hematuria, ruptura y sangrado. Presentan un alto riesgo del desarrollo de hipertensión e insuficiencia renal crónica, debido a la compresión y la sustitución del parénquima renal.13
La característica cardiovascular más común en el CET es el rabdomioma que se manifiesta en 50 - 70 % de los casos, es un tumor benigno, asintomático, localizado en los ventrículos; pueden provocar arritmias cardíacas incluyendo arritmias atriales y ventriculares e insuficiencia cardíaca. Es una de las primeras manifestaciones del CET pudiendo detectarse intrautero.2, 8, 14
El compromiso pulmonar en el CET está presente en 1% a 6%, entre la más característica se encuentra la linfangioleiomiomatosis en 1 - 3 %, afecta de forma exclusiva a las mujeres entre la 3° y 5° década de la vida y es debido a la acción reguladora de los estrógenos sobre vías de señalización celular implicadas en el CET. Con menor frecuencia la hiperplasia micro nodular multifocal de neumocitos, quistes pulmonares y tumores de células claras del pulmón y pueden dar lugar a disnea, cor pulmonale y neumotórax espontáneo.
Entre los hallazgos oculares se observa con mayor frecuencia los hamartomas astrocíticos de la retina en un 47 - 87 %. Algunos pacientes manifiestan perdida visual, pero la ceguera es rara. Otras manifestaciones son angiofibromas de los párpados, colobomas, estrabismo y pérdida de la pigmentación sectorial del iris.
Las lesiones óseas están presentes en un 50 % de los casos, siendo más frecuentes los quistes óseos localizados a nivel de falanges, metacarpo y metatarso; neoformación ósea perióstica, áreas de esclerosis en columna, pelvis y cráneo. Las depresiones puntiformes del esmalte dentario se observan en el 70-100% de los pacientes, son irregulares, de 1 a 2 mm de diámetro, presentes tanto en dientes deciduos como en los permanentes.
Con menor frecuencia se pueden hallar pólipos rectales, generalmente hamartomatosos y pocas veces adenomatosos, afectación hepática, esplénica, pancreática, hamartomas en tiroides, suprarrenales y síndromes endocrino-metabólicos.
El diagnóstico diferencial variará y dependerá de las manifestaciones clínicas de cada caso. Algunas manifestaciones clínicas pueden aumentar la sospecha de complejo esclerosis tuberosa, incluidos rabdomiomas cardiacos prenatales, convulsiones infantiles, maculas de piel hipopigmentadas y trastornos del espectro autista.3
Algunas manifestaciones de CET son inespecíficas y pueden aparecer de forma esporádica en individuos normales o ser marcadores de otras enfermedades. Cuando las manifestaciones clínicas típicas de CET aparecen de forma aislada o en grupo, pero sin cumplir suficientes criterios para el diagnóstico de certeza de la enfermedad y con un estudio genético de TSC1/TSC2 negativo o no concluyente (por ejemplo, una variante de significado incierto), suele tratarse de formas en mosaico. No obstante, es importante descartar otras enfermedades que pueden presentar algunas manifestaciones de CET, como el síndrome de neoplasia endocrina múltiple tipo 1 (MEN1) en caso de angiofibromas faciales, sobre todo si aparecen en edad adulta o el síndrome de Birt-Hogg-Dubé en caso de quistes pulmonares.15, 16
Dentro de los diagnósticos diferenciales de las máculas hipomelanóticas se encuentran: nevo anémico, nevo acrómico, piebaldismo, vitíligo e hipopig-mentación post inflamatoria.4
La piel de zapa se puede ver también en nevos de colágeno congénito o adquirido incluyendo colagenoma cutáneo familiar, colagenomas eruptivos y colagenoma aislado.4
Los fibromas ungueales o periungueales (tumores de Köenen) se pueden confundir con fibroqueratoma acral.4
Para realizar un buen tratamiento es indispensable realizar un buen examen físico y de esta manera un diagnóstico certero. El tratamiento debe ser multidisciplinario, incluyendo asesoría genética, la cual es útil para pacientes y padres de niños con CET.
Se debe informar a los padres que el riesgo de tener hijos que padezcan esta enfermedad en caso de mutación en TSC1 o TSC2 ó en caso de No Mutación Identificada (NMI) en TSC1 o TSC2 en el estudio molecular es de un 50 % de los casos.3,5, 6, 7
El 2/3 de los casos son esporádicos o mosaicismo y 1/3 de los pacientes tiene un progenitor afectado.
Es de vital importancia descartar la enfermedad en los padres sin manifestaciones clínicas, realizando un análisis minucioso de mutaciones de los genes implicados a través de un estudio molecular y un adecuado examen físico incluyendo exploración de la piel, retina, RM cerebral y ecografía renal. Si los estudios realizados son negativos para CET se considera un mosaicismo o caso esporadico en uno de los progenitores.18 Los Padres que no están afectados por la enfermedad tienen el riesgo de padecer de este en un 1 - 2 %.3, 17
El riesgo de que los hermanos padezcan la enfermedad depende del estudio realizado en los padres, si existe una mutación el riesgo de que los hermanos padezcan la enfermedad es del 50 %, si los padres no presentan la enfermedad y es un mosaicismo el riesgo de que los hermanos padezcan la enfermedad es del 3 %.18
La evaluación dermatológica es útil para el reconocimiento de lesiones precoces de la piel tales como los angiofibromas y los fibromas ungueales (tumores de Köenen), capaces de causar desfiguraciones estéticas, y el posterior requerimiento de tratamientos quirúrgicos que incluyen, cirugía convencional, criocirugía, terapia láser y dermoabrasión, con un pronóstico empobrecido por las altas tasas de recidivas de estas lesiones”. 3, 4, 15
Las máculas hipomelanóticas pueden ocultarse temporalmente mediante cremas autobronceadoras o maquillaje del color de la piel del paciente. Las placas de la piel de zapa, por lo general no se tratan, aunque pueden extirparse.1
Al igual que en la piel, se recomienda la rápida intervención de Odontología, de preferencia, profesionales con experiencia en el tratamiento de esta variedad de lesiones orales, particularmente las sintomáticas, por su tendencia a aumentar tanto en número como en tamaño, capaces entonces de causar compromiso funcional.17
Se ha evidenciado en estudios mejoría de astrocitomas subependimarios de células gigantes, reducción de angiomiolipomas renales y mejoría de la función pulmonar en los pacientes con linfangioleiomiomatosis, e incluso de angiofibromas con el uso de rapamicina vía oral, pero es probable que las reacciones adversas graves limiten la administración vía oral en lesiones de la piel, sobre todo en niños.4, 17
Se ha informado mejoría de angiofibromas con rapamicina tópica, pero los enfoques quirúrgicos siguen siendo el pilar del tratamiento.4, 17
La supervivencia del CET depende de la gravedad de síntomas neurológicos y del desarrollo de insuficiencia renal, por lo tanto, el seguimiento de estos pacientes debe ser exhaustivo y de por vida por todas las especialidades involucradas para detectar nuevas lesiones prevenibles y tratables.19
CONCLUSIONES
El interés de la publicación del caso es presentar un paciente adulto con un diagnóstico tardío de CET, quien fue tratado como síndrome convulsivo desde la infancia, cumpliendo con la tríada clínica característica, así como los criterios clínicos. Nuestro paciente presentó cuatro criterios principales y tres criterios secundarios, incluyendo dentro de estos los tumores de Köenen gigantes en ambos pies, los angiofibromas faciales y el signo de piel de zapa en región lumbosacra.
Es importante conocer la existencia de este síndrome para poder diagnosticarlo en la consulta médica. Además es vital resaltar la relevancia de un examen físico completo para arribar a un diagnóstico precoz y una intervención oportuna.
La evaluación dermatológica ayuda a realizar un diagnóstico temprano para un tratamiento oportuno y precoz, brindando una mejor calidad de vida, evitando posibles complicaciones.
Por último, dejamos en evidencia la evolución inusual del caso de un paciente adulto con CET con la presencia de complicaciones de tumores de Köenen gigantes que causaron alteración y dolor a la marcha, con buena respuesta al tratamiento quirúrgico por el servicio de cirugía plástica, teniendo en cuenta la importancia de la coordinación multidisciplinaria incluyendo el consejo genético tanto en el paciente como en los familiares.

