











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El granuloma actínico (GA) fue descripto por primera vez en 1975 por John O’Brien. Es una rara dermatosis granulomatosa caracterizada por placas anulares con atrofia central y márgenes eritematosos elevados, localizadas en áreas fotoexpuestas. Afecta a personas de mediana edad con antecedentes de exposición solar intensa.1
El GA es un trastorno relativamente raro y se desconoce su patogenia exacta.
Dentro de los diagnósticos diferenciales a tener en cuenta se encuentran: tiña corporis, sarcoidosis y granuloma anular. El diagnóstico se confirma con la histopatología. Aunque no existe un tratamiento específico para GA, se han probado varios agentes terapéuticos con variados resultados.
CASO CLÍNICO
Paciente femenino de 71 años de edad, hipertensa, que consulta por dermatosis ligeramente pruriginosa compuesta por placas eritematosas con fina escama en su superficie, de configuración anular, de bordes elevados, con centro levemente atrófico; por sectores confluyen formando placasmás grandes, localizadas en ambos antebrazos y dorso de manos (Figura 1). La paciente trae un estudio micológico con resultado positivo para Microsporum. Se inició tratamiento con fluconazol 150 mg por semana, durante 4 semanas asociado con clotrimazol tópico. No se observó mejoría clínica.
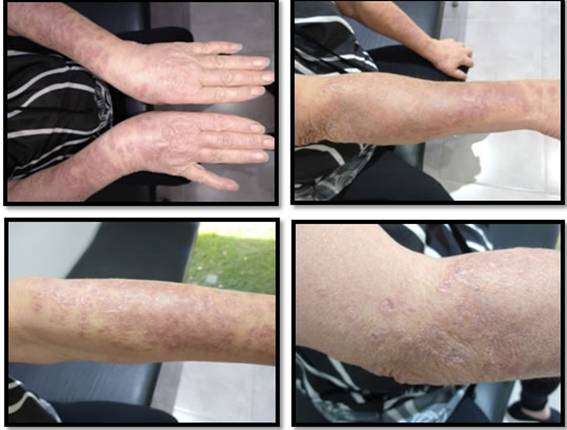
Figura 1: Placas eritematosas anulares con fina escama, bordes elevados, con centro atrófico, formando placas grandes en antebrazos y dorso de manos.
Se realizó una biopsia por punch para estudio histopatológico que mostró a nivel dérmico reacción granulomatosa dérmica constituida por numerosas células gigantes multinucleadas, sueltas y agrupadas (sin corona linfocitaria). No se observa necrosis caseosa (Figura 2). Con técnica especial de Grocott no se aprecian elementos micóticos. Con técnica de Orceína para fibras elásticas se aprecia elastofagocitosis en células gigantes (Figura 3).
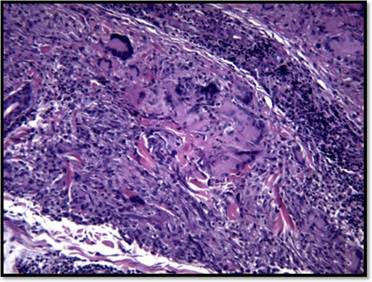
Figura 2: HE-10x: A nivel dérmico reacción granulomatosa dérmica compuesta por células gigantes multinucleadas sin corona linfocitaria ni necrosis caseosa.
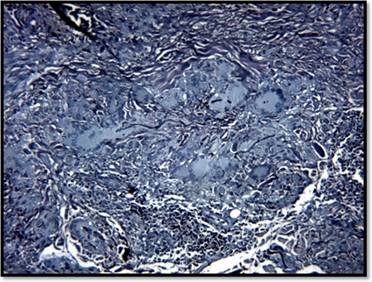
Basados en la clínica y la histopatología se arribó al diagnóstico de granuloma actínico. Se inicia tratamiento conclobetasol ungüento semana por medio e infiltraciones con triamcinolona mensuales, junto con fotoprotección estricta, observándose buena respuesta clínica, sin progresión de las lesiones (Figura 4).
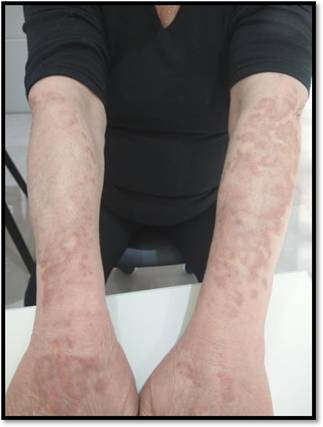
DISCUSIÓN
El granuloma actínico (GA) fue descrito por J.P. O'Brien en 1975 como un fenómeno de reparación del tejido conectivo elastótico dañado por el sol y/o el calor. Realizó una minuciosa descripción histológica, afirmando que se trata de una entidad relacionada con el granuloma anular, pero con características propias que permiten distinguirlo de otros granulomas, presentándolo como una entidad independiente. El granuloma anular nomuestra elastofagocitosis y sí presenta depósitos de mucina,necrobiosis y granulomas en empalizada.2
Desde entonces ha recibido múltiples denominaciones, tales como granuloma de O’Brien,necrobiosis lipoídica atípica de la cara y del cuero
cabelludo,granuloma disciforme de la cara deMiescher,granulomamultiforme, entre otros.
Es un trastorno relativamente raro que se observa con mayor frecuencia en adultos de mediana edad, de piel clara, con antecedentes de exposición excesiva al sol. Tiene un ligero predominio por el sexo femenino. Su prevalencia actual sigue siendo incierta.
Se desconoce la patogenia de la enfermedad sin embargo se cree que la elastosis solar es un factor desencadenante.3Las fibras elásticas dañadas se consideran como los estímulos antigénicos que desencadenan el proceso inflamatorio humoral y celular, que conduce a la reparación del tejido conectivo dañado por el sol.
En 1979 Henke et al., describieron al Granuloma elastolítico anular de células gigantes (GEACG)como un cuadro clínicoe histológicamente similar, salvo por no estar limitada a áreas de exposición solar.4
Tanto el granuloma actínico como el granuloma anular elastolítico de células gigantes correspondería, de acuerdo a las diversas bibliografías, a una misma entidad salvo por el detalle que las lesionesclínicas no se limitan a áreas fotoexpuestas, pero su histopatología no difiere.No son entidades diferentes como exponen los autores.5
El GA se presenta típicamente como placas anulares con márgenes eritematosos elevados y atrofia o hipopigmentación central, en cara, cuello, pecho y extremidades superiores. Las lesiones pueden iniciar como pápulas eritematosas y luego expandirse de manera centrífuga y fusionarse en placas anulares o lineales asintomáticas.1,6
El diagnóstico diferencial incluye: Granuloma anular clásico, Tiña corporis, Liquen anular, Sarcoidosis, y otras enfermedades granulomatosas infecciosas, pero se pueden distinguir fácilmente por los hallazgos histopatológicos y de laboratorio.
El estudio histopatológico del GA muestra un proceso inflamatorio granulomatoso limitado a la dermis superficial. El rasgo histopatológico característico del GA son las células gigantes multinucleadas de tipo cuerpo extraño, que fagocitan las fibras elásticas degeneradas, un proceso conocido como elastofagocitosis.7
Se han descripto cuatro patrones histológicos de GA: Histiocítico, de
Células gigantes, Necrobiótico o vascular y Sarcoidal.
La variante histiocítica consiste predominantemente en histiocitos dispersos cerca de fibras elásticas. El patrón de células gigantes es el patrón original, que inicialmente consiste en un pequeño granuloma que se vuelve anular a medida que avanza hacia el tejido elastótico cercano. La variante necrobiótica o vascular, consiste en áreas de necrosis isquémica en el borde granulomatoso que avanza. Por último, la variante sarcoide a menudo muestra estroma atrófico, fibrosis y una reacción inflamatoria persistente. De los cuatro patrones, el de células gigantes y necrobiótico parecen ser patrones más comunes en mujeres que en hombres.3
En el caso de la paciente presentada, la histopatología correspondería a un patrón de GA de células gigantes.
El tratamiento del GA incluye: corticosteroides tópicos e intralesionales, terapia PUVA, antipalúdicos, ciclosporina, metotrexato y crioterapia. Los informes de casos y los estudios observacionales han demostrado resultados variables con estas modalidades de tratamiento.8-12
Existe un reporte de caso en el cuál un paciente fue tratado con tetraciclinas obteniéndose buena respuesta clínica.9Se ha informado remisión espontánea en algunos casos.

