











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
El ganglioneuroma suprarrenal (GnS) es un tumor benigno, raro y de crecimiento lento, originado a partir de las neuronas simpáticas primitivas de la cresta neural. Puede surgir en cualquier lugar a lo largo del plexo simpático paravertebral, más frecuentemente en mediastino posterior y retroperitoneo. La localización adrenal ocupa el menor porcentaje y se han reportado menos de 25 casos en la literatura™.
CASO CLÍNICO
Paciente femenina de 37 años de edad, sin antecedentes patológicos relevantes consulta por dolor intermitente en fosa renal izquierda y flanco ipsilateral de 5 meses de evolución, sin otros síntomas acompañantes. Al examen físico, se encuentra normotensa, con frecuencia cardíaca de 80 latidos por minuto, sin signos sugestivos de síndrome de Cushing o hiperandrogenismo clínico. Se realiza ecografía abdominal que informa masa homogénea, hipoecoica, de limites definidos, adyacente al riñón izquierdo, en su porción superior, de 80 x 50 x 70 mm. La TAC abdominopélvica informa lesión hipodensa en fase sin contraste, con densidad tisular de 30 UH, en fase con contraste presenta washout del 40% con refuerzo heterogéneo, áreas de necrosis, refuerzos nodulares murales y calcificaciones en región inferior. La resonancia magnética contrastada identifica formación expansiva solida, oval, de 85 x 58 x 74 mm,de contornos definidos, con señal hipointensa y homogénea en T1 y discretamente hipointensa y heterogénea en T2, con refuerzo máximo al contraste, en forma heterogénea, lavado lento y sin caída de señal fuera de fase. Se continuó con estudio hormonal completo para descartar funcionalidad de la lesión (Figura 1).
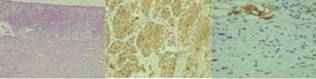
Figura 1: a- Las secciones histológicas muestran restos de parénquima de glándula suprarrenal, con una proliferación de bordes bien delimitados, netos, constituida por células fusadas, con núcleos elongados y citoplasmas eosinófilos, dispuestas en haces entremezclados, con aisladas células ganglionares. b- Las células ahusadas descriptas (células de Schwann) exhibieron positividad difusa con S100, confirmando estirpe neural. c- Se observaron aisladas células ganglionares, con citoplasma eosinófilo granular amplio, positivas con cromogranina y sinaptofisina.
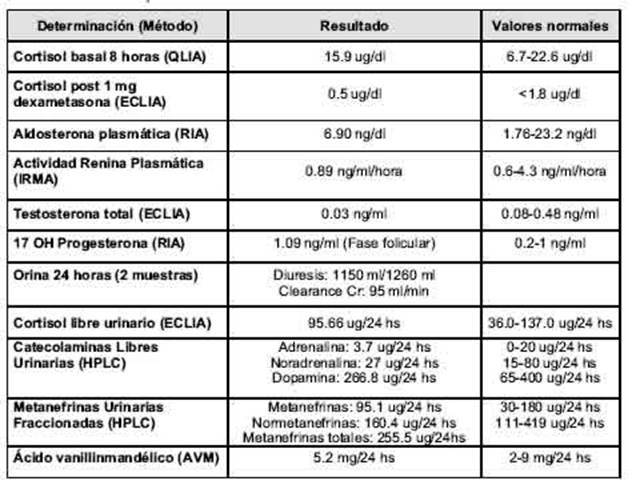
Las determinaciones de laboratorio realizadas fueron: cortisol basal, cortisol libre urinario de 24 horas, Test de Nugent, Actividad Renina Plasmática (ARP) y aldosterona plasmática, catecolaminas y metanefrinas urinarias, ácido vainillilmandélico (AVM), testosterona total y 17 OH progesterona, las cuales resultaron normales, descartando hipercortisolismo, hiperaldosteronismo, feocromocitoma funcionante e hiperandrogenismo (Tabla 1)
Dadas las características imagenológicas y el tamaño de la lesión, se decide tratamiento quirúrgico, considerando a feocromocitoma silente como principal diagnóstico presuntivo pre quirúrgico. Se realizo adrenalectomía laparoscópica con abordaje por lumbotomía, con resección total del tumor. La evolución postoperatoria fue excelente. El informe de anatomía patológica evidenció formación nodular de 83 x 79 x 69 mm, de superficie lobulada, rosada, con escaso tejido adiposo adherido. Al corte, el tejido es firme y arremolinado. Al examen microscópico, se observa proliferación nodular, de bordes delimitados, formado por células fusadas, con núcleos bipolares y citoplasmas elongados, correspondiendo a células de Schwann con células ganglionares aisladas. En la inmunomarcación presenta aisladas células ganglionares con tinción positiva para Cromogranina A y Sinaptofisina, S100 positivo difuso, índice de proliferación Ki67 del 4%, se descarta componente asociado de feocromocitoma, hallazgos que resultan consistentes con ganglioneuroma suprarrenal.
DISCUSIÓN
Los tumores neuroblásticos se originan en las células de la cresta neural del tejido nervioso simpático y pueden formarse en cualquier punto del sistema paravertebral y la médula suprarrenal. La clasificación actual incluye: neuroblastomas, ganglioneuroblastomas y ganglioneuromas, los cuales se diferencian por su grado de maduración. Esto se relaciona con su pronóstico siendo a mayor inmadurez, mayor su agresividad. El ganglioneuroma es un tumor benigno, infrecuente, de crecimiento lento, que deriva de la cresta neural, compuesta por células ganglionares maduras (totalmente diferenciadas) y células mesenquimales de Schwann1-2. Afectan principalmente a niños y adultos jóvenes. La mayor parte de estos tumores se sitúan en el mediastino posterior (40%) y retro peritoneo (37%), la localización adrenal se estima en un 15-30%. Su tamaño es variable, con un promedio de 8 cm. Su detección suele ser incidental, dado que generalmente se mantienen hormonalmente inactivos; aunque en un 20-30% de los casos pueden producir catecolaminas y metabolitos, incluso de manera subclínica. Los signos y/o síntomas más frecuentes son: dolor abdominal, lumbar inespecífico o masa palpable y ocasionalmente se pueden asociar a diarrea (debido a liberación de péptido intestinal vasoactivo), sudoración e hipertensión®. En relación a las características por imagen, la ecografía, tomografía computada y resonancia magnética reportan una masa bien definida, oval o lobulada. La ecografía suele evidenciar una masa homogénea e hipoecoica, de límites definidos. La tomografía sin contraste muestra tumor homogéneo, hipodenso con bajos índices de atenuación (10-40 UH), en un pequeño porcentaje puede observarse calcificación fina y moteada. Además, puede presentar refuerzo heterogéneo del contraste con lento washout en fase tardía, que fue el caso de nuestra paciente. De ahí la importancia del diagnostico diferencial con otro tipo de tumores como carcinoma adrenal o feocromocitoma, ya que tanto el tamaño como las características imagenológicas pueden ser similares. La resonancia magnética los presenta como lesiones con señal homogénea baja en T1 y señal heterogénea e hiperintensa en T2, pueden presentar ausencia de caída de señal en fase opuesta, característico de lesiones no adenomatosas*3®. Luego de la caracterización por imagen, el proceso diagnóstico continua con el estudio hormonal, que demuestre la funcionalidad o no del tumor con determinaciones de cortisol basal 8 horas, cortisol libre urinario de 24 horas, Test de Nugent, Actividad Renina Plasmática, aldosterona plasmática, catecolaminas y metanefrinas urinarias, testosterona total y 17 OH progesterona. El diagnóstico definitivo se realiza con el estudio histopatológico e inmunohistoquímico de la pieza quirúrgica. El estudio microscópico muestra estroma constituido por células de Schwann, con característica fusiforme, escaso citoplasma y núcleo oscuro, que se entrecruzan en forma irregular. Dispersas en este fondo se encuentran células ganglionares maduras, con citoplasma eosinófilo, formando pequeños grupos. En el estudio por inmunohistoquímica presenta positividad para marcadores neuroendócrinos como cromogranina A (CgA), sinaptofisina y proteína S1004. En relación a la conducta quirúrgica en tumores adrenales no funcionantes, la Guía de Práctica Clínica de Manejo de Incidentalomas Adrenales de la Sociedad Europea de Endocrinología sugiere adrenalectomía laparoscópica en masas adrenales con hallazgos radiológicos sospechosos de malignidad y menores o iguales a 6 cm, sin evidencia de invasión local (R. 4.3) y recomienda adrenalectomía abierta en cuando existen signos de invasión (R. 4.4)(5-6) En nuestro caso, se decidió exéresis de la misma por el tamaño y las características sospechosas por imagen. Se realizó adrenalectomía laparoscópica por lumbotomía, ya que la lesión presentaba márgenes definidos sin signos de invasión. Este abordaje se asocia a menor tiempo quirúrgico, menor estancia hospitalaria y riesgo de complicaciones. El pronóstico del GnS tras la resección completa es muy bueno y la recurrencia es excepcional.
CONCLUSION
Si bien la forma de presentación de este caso, con síntomas inespecíficos y hallazgo incidental de masa adrenal es la más frecuente descripta en la literatura, estamos ante un caso atípico de Gn, dado que es un tumor infrecuente y solo un mínimo porcentaje se presenta en la glándula suprarrenal. Por lo tanto, ante el hallazgo de una masa adrenal mayor de 4 cm, bien delimitada, con calcificaciones puntiformes, índice de atenuación entre 10-40 UH, con signos de lesión atípica, sin producción hormonal asociada, debe considerarse el diagnostico de GnS como parte del estudio de las lesiones incidentales. A pesar de los datos que nos brindan los estudios de laboratorio (no funcionante o con producción hormonal subclínica) o por imagen, este tipo de lesiones comparten características estructurales con tumores potencialmente malignos, por lo que es importante plantear el estudio exhaustivo y el tratamiento quirúrgico con posterior confirmación histopatológica, sobre todo si se presenta en niños o adultos jóvenes, característico de este tipo de neoplasias, dado el excelente pronostico que presenta y la baja tasa de recidiva luego del tratamiento.

