











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkEl término encefalopatías epilépticas ha incluido varias entidades que cursan con frecuente actividad epileptifor me interictal que típicamente causa un impacto adverso en el desarrollo, causando enlentecimiento o regresión cognitiva y del comportamiento más allá de lo esperado para las lesiones de base1. En forma reciente la Liga Inter nacional contra la Epilepsia (ILAE) socializó la propuesta de clasificación, definición y nomenclatura para designar los síndromes epilépticos de inicio en el período neona tal y los primeros dos años de vida2, la cual incluye los síndromes autolimitados y las encefalopatías epilépticas y del desarrollo (DEEs). El concepto de encefalopatía epiléptica y del desarrollo contempla que los niños se presentan con una epilepsia grave de inicio temprano, con compromiso del desarrollo que podría ser atribuido tanto a la etiología como a los efectos adversos de la actividad epiléptica no controlada2.
Los pacientes pueden presentar diferentes tipos de crisis incluyendo estado epiléptico y aunque algunas encefalopatías podrían no tener una alta tasa de eventos ictales, es frecuente que el deterioro sea mayor en perío dos de frecuentes crisis con resistencia a los tratamientos farmacológicos3,4.
Muchos de los pacientes tiene causas estructurales y metabólicas pero los avances recientes en genética han permitido encontrar bases moleculares que permiten di rigir mejor el tratamiento. Es fundamental no solo detener las crisis para evitar el deterioro sino conocer la etiología, evitando medicamentos que pueden empeorar un cuadro como en el caso de la carbamazepina y la oxcarbazepina en mutaciones con pérdida de la función del canal de sodio que se presenta en la mutación de la subunidad alfa 1 del canal del sodio SCN1A en el caso del síndrome de Dravet y de ésta seleccionar medicamentos como el stiripentol o la fenfluramina3; pero además considerar el uso de los mismos bloqueadores de sodio, carbamazepina u oxcarbazepina en la encefalopatía temprana neonatal donde la mutación en el gen SCN1A genera una ganancia de la función.
La evolución y pronóstico de estas entidades depende de la edad de presentación, de las comorbilidades, de la rapidez con que seleccionemos el tratamiento adecuado y especialmente de la etiología; de esta manera podría mos hacer medicina de precisión, eligiendo el mejor medicamento anti-crisis para cada paciente, evitando algunos, supliendo un error metabólico o identificando tempranamente lesiones estructurales indicativas de cirugía de epilepsia que mejoren el pronóstico y el curso de estas enfermedades.
En la presente revisión nos dedicaremos a las en cefalopatías epilépticas de presentación en el neonato y el lactante puntualizando los hallazgos clínicos, electroencefalográficos, imagenológicos y metabóli cos, así como los avances moleculares y tratamientos recomendados.
Clásicamente han sido conocidas como encefalopatías con patrón de “estallido - supresión” de inicio en los prime ros 3 meses de vida1, caracterizadas por pobre respuesta a los tratamientos antiepilépticos, alta mortalidad y grave compromiso del desarrollo en los que sobreviven5.
En la reciente clasificación de la ILAE se han de nominado “Encefalopatías epiléptica y del desarrollo infantil temprano (EIDEE)” agrupando el espectro de las reconocidas epilepsia mioclónica temprana (EMT) y la encefalopatía epiléptica infantil temprana o Síndrome de Ohtahara2,6 debido a que ambos síndromes comparten el mismo patrón electroencefalográfico, edad de inicio, pronóstico así como tipos de crisis aunque predominen las mioclonias en la primera y las crisis tónicas en el Sín drome de Ohtahara; las etiologías estructurales en el S. de Ohtahara y las metabólicas en la EMT5. Los criterios diagnósticos incluyen2:
- Inicio de la epilepsia en los primeros 3 meses con crisis típicamente resistentes al tratamiento
- Examen neurológico anormal, alteraciones de la postura, tono o movimiento
- Compromiso del desarrollo moderado a profundo evidente con el tiempo
- Electroencefalograma interictal anormal con patrón estallido supresión, enlentecimiento difuso o descargas multifocales
- Estudios metabólicos, neuro-imagenológicos o genéticos esclarecen la etiología en aproximadamente 80% de los casos
Dentro de las causas metabólicas podemos encontrar hiperglicinemia no cetósica, deficiencia de co-factor de molibdeno, deficiencia de sulfito oxidasa, acidemiame tilmalónica y propiónica, déficit de piridoxina, déficit de PNPO (piridoxina 5’-fosfato oxidasa) y enfermedad de Menkes, entre otros.
Cada vez se reconocen más genes involucrados en estos trastornos7,8, por ejemplo, el gen STXBP1 asociado al síndrome de Ohtahara en el 30% de los casos, KCNQ2 en el 20%, SCN2A en el 10%. Algunos genes podrían correlacionarse con un tipo de crisis, como las variantes pa togénicas de los genes KCNQ2, SCN2A, STXBP1, KCNT1 con crisis secuenciales y crisis tónicas, componente auto nómico, SCN8A con crisis focales,CDKL5 con espasmos tónicos hipercinéticos y UBA5 con crisis mioclónicas2.
El diagnóstico de EIDEE requiere una o más de las siguientes crisis: mioclonías, espasmos, crisis tónicas y crisis secuenciales2, las cuales ocurren incluso desde el primer día de vida. Los espasmos epilépticos y las crisis tónicas pueden ocurrir en salvas con un componente focal o asimétrico y también pueden presentarse crisis focales comprometiendo solo un hemicuerpo. Muchas pueden correlacionarse con alteración estructural5.
Las mioclonias son más frecuentes en la EMT y pue den ser axiales, segmentarias o erráticas, éstas últimas pasando de la cara a una extremidad. Es infrecuente que sean generalizadas.
Con frecuencia crisis focales tónicas o clónicas se preceden o se siguen de una miocloníao una crisis tónica asimétrica que podrían iniciar o terminar con un componente autonómico o apnea y desaturación; este tipo de crisis se ha denominado crisis secuenciales en la última clasificación de crisis neonatales de la ILAE6 y pueden verse también en síndromes autolimitados neo natales o enencefalopatías asociadas con mutaciones en KCNQ2, KCNQ3; que pueden beneficiarse del uso de carbamazepina6,7.
Inicialmente los registros pueden ser normales, pero rápidamente se pierde el patrón esperado para la edad, con pérdida de la organización y aparición del patrón estallido supresión que en el síndrome de Ohtahara se presenta tanto en la vigilia como en el sueño y en la EMT es más notorio durante el sueño (Fig. 1). También puede observarse el compromiso de un solo hemisferio (hemiestallido) relacionado con lesiones estructurales como hemimegalencefalia, esquicenzefalia, síndrome de Aicardio actividad paroxística interictal focal sobrepuesta.
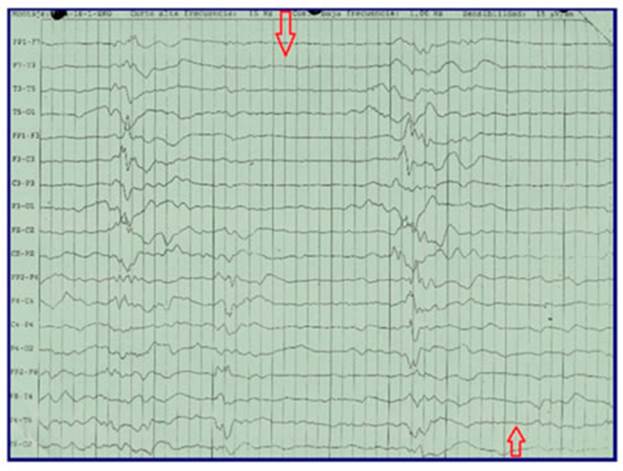
El patrón de estallido supresión consiste en brotes de actividad delta o theta de alto voltaje asincrónicos mez clados con puntas o polipuntas de hasta 150 a 350 uVol de amplitud, de 1 a 6 segundos de duración que alternan con períodos de quiescencia o supresión de 2 a 5 o hasta 18 segundos de duración de menos de 10 uVol9,10.
La refractariedad al tratamiento es común a menos que se tenga una etiología estructural quirúrgica, desórdenes metabólicos o genéticos tratables como el déficit de Glut 1 que responde a dieta cetogénica. Pacientes con variantes patogénicas en SCN2A o SCN8A podrían responder a dosis altas de bloqueadores de canales de sodio2.
La epilepsia dependiente de piridoxina (ALDH7A/ Antiquitin) responde a la administración de vitamina B6 100 mg seguidos de 15-30 mg/k/d. La epilepsia depen diente de piridoxal 5’fosfato PLP (PNPO), a 30 mg/k/d de PLP dividido en 3 dosis y las crisis respondedoras a ácido folínico (FARS), a 0.5-1 mg/k/d de ácido folínico con dosis de mantenimiento de 2-5 mg/k/d.
Clásicamente se presenta en los primeros 6 meses de vida en niños sin antecedente de noxa perinatal, inicial mente con crisis focales motoras clónicas aisladas, con o sin componente autonómico con correlación electroence falográfica topográfica caracterizada por actividad rítmica theta que posteriormente va migrando a otra región, asociado a un enlentecimiento difuso de la actividad de base1. Las crisis se hacen cada vez más frecuentes y son refractarias al tratamiento asociadas a estado epiléptico. Se han descrito crisis tónicas focales o multifocales, eventualmente espasmos pero las mioclonias son crite rio de exclusión2. Una videoelectroencefalografia podría evidenciar mejor el patrón migratorio.
Causa un deterioro marcado y progresivo del desarrollo con pérdida de habilidades adquiridas, hipotonía, micro cefalia y falta del contacto visual, trastornos de motilidad gástrica y movimientos anormales. Los pacientes tienen imágenes de resonancia cerebral normales o con hallaz gos sutiles y estudios metabólicos normales o aislados casos de defectos de glicosilación y con frecuencia no tienen antecedente familiar. Fue descrita inicialmente en 199511 y durante muchos años se desconoció su causa. Se reconocen ahora más de 30 genes asociados: KCNT1, SCN2A, SCN1A, PLCB1, GABRB3, HCN1, SCN8A, SMC1A, BRAT1, SLC12A5, SLC25A22, ITPA y KARS, entre otros8,12.
La mutación más común es en el gen del canal de po tasio KCNT1, que se ha relacionado con otros fenotipos, como en epilepsia frontal nocturna autosómica dominante. Todas las mutaciones en KCNT1 asociadas a epilepsia determinan ganancia de función.
Se denomina en forma más amplia para incluir pacientes que presentan espasmos epilépticos aislados que no cum plen la triada clásica del síndrome de West: espasmos in fantiles, retardo en el desarrollo psicomotor e hipsarritmia mencionada por el Dr. West en su descripción inicial5,13 y luego descrita en 1951 por Vázquez y Turner quienes la denominaron “epilepsia en flexión generalizada”.
Es una epilepsia edad dependiente que ocurre entre 1 y 24 meses, predominantemente en lactantes, con un pico entre los 3 y 7 meses, se deben considerar otras en cefalopatías si inician antes de los 3 meses. Clínicamente está caracterizada por la presencia de espasmos de tipo flexor, extensor o mixtos frecuentemente en salvas2,13. Los episodios al inicio y en los pacientes parcialmente tratados pueden ser sutiles y mostrarse solo como una pequeña supraversión de mirada; característicamente ocurren en salvas y pueden tener un componente focal. Los padres refieren con frecuencia pérdida del contacto visual de manera inicial.
En el 55% de los casos se aprecian lesiones estructu rales por noxa perinatal, malformación cerebral, anorma lidades del desarrollo cortical, infecciones intrauterinas, síndromes neurocutáneos - el más frecuente, la esclerosis tuberosa -; rara vez los pacientes tienen antecedente familiar de epilepsia.
El pronóstico está relacionado con la etiología y el reconocimiento temprano, así como su tratamiento ade cuado y oportuno. Los pacientes sin alteración estructural tienen un mejor pronóstico2,5.
Cuando solo contamos con EEG basal y no con video electroencefalografía debemos tener en consideración que los hallazgos electroencefalográficos, especialmente al inicio del cuadro, ocurren durante el sueño no REM y los registros de vigilia y de sueño REM pueden ser nor males o en ocasiones muestran actividad interictal focal. Los espasmos generalmente aparecen al despertar, por lo tanto, la grabación debe continuar por lo menos 15 minutos después del alertamiento14.
El uso de electrodos adicionales de EMG en deltoides es muy útil, en términos de reconocer el espasmo y dife renciarlo de mioclonias y convulsiones tónicas.
La mayoría de los espasmos epilépticos tienen una contracción fásica de 1 a 2 segundos con correlación elec tromiográfica en forma de rombos o signos de mitad de rombo. En mioclonías se observan breves contracciones de menos de 1 segundo sin morfología típica. En convul siones tónicas se observa una contracción sostenida y homogénea durante más de 2 segundos.
El patrón de hipsarritmia clásico, una vez instaurado el cuadro, consiste en un patrón caótico que desorga niza el trazado de fondo. En vigilia se aprecia actividad de alto voltaje de puntas y ondas lentas multifocales o focales que pueden variar de localización y duración con un predominio posterior pero que nunca es rítmica5. En sueño no REM se fragmenta adquiriendo un patrón pseudoperiódico (Fig. 2) y desaparece en sueño REM. Se han descrito diferentes variantes de hipsarritmia como la hipsarritmia con incremento de sincronización interhemis férica, hipsarritmia asimétrica, hipsarritmia con episodios de atenuación de voltaje, hipsarritmia con componente focal y otros14,15.

Ictalmente se distinguen 3 patrones: una onda lenta trifásica de alto voltaje difusa, una descarga rápida de bajo voltaje, o un aplanamiento difuso de la actividad de corta duración. Los hallazgos tanto ictales como interic tales pueden ser asimétricos cuando existe una lesión estructural.
Se han encontrado varias mutaciones en estudio de pacientes sin noxa clara, destacando STXBP1, ARX (en varones), CDKL5 (en niñas), SPTAN1, SCN1A, GRIN2A, GRIN 2 B, STXB1, SLC2A1, CASK, ALG13, PNPO, CDKL5, MAGI2, SCN1A, SCN2A, STK9, DCX y ADXL8.
El tratamiento hormonal (con corticotropinaintramus cular o corticoides orales) y la vigabatrinason la primera elección, pero la duración y dosis de cada uno varían entre guías. Nosotros usamos el protocolo sugerido por Hussain y col.16 frecuentemente combinado con vigaba trina, este último de elección en esclerosis tuberosa17. Otros tratamientos, considerados de segunda línea, incluyen ácido valproico, topiramato, benzodiacepinas, levetiracetam, rufinamida, zonisamida, dieta cetógeni ca y piridoxina. Es fundamental actuar rápido y tomar conductas en corto tiempo intentando control completo clínico y electroencefalográfico para mejorar el pronóstico siguiendo algoritmos como el último propuesto en la revisión de Iype y Koshy17.16
Inicialmente conocida como epilepsia mioclónicagrave de la infancia; sin embargo, las mioclonias no son su mani festación inicial y se han descrito casos sin mioclonias. Sus primeras manifestaciones ocurren en el primer año de vida con crisis convulsivas generalizadas, unilaterales o focales asociadas a fiebre, frecuentes o recurrentes y prolongadas. Disminuyen en duración con el tiempo y en el segundo o tercer año de vida se presenta mioclonus masivo con cambios electroencefalográficoso mioclonu serrático usualmente fragmentario no asociado a cambios electroencefalográficos. Posteriormente persisten las crisis frecuentes de mioclonias, focales o de ausencias atípicas, asociados a deterior cognitivo y del lenguaje.
El EEG inicialmente puede ser normal, a pesar de la frecuencia de las crisis convulsivas. Hacia el segundo año aparecen complejos de punta onda o polipunta onda lenta rápida aislados o en brotes generalizados, focales o multifocales. La fotoestimulación es positiva en el 40% de los pacientes18.
Con los años se aprecian puntas multifocales u ondas agudas que aumentan durante el sueño lento y el trazado de fondo se hace lento. Sobre el vértex y regiones cen trales se puede observar ritmo theta de 5-6cps desde el inicio.
Se encuentran mutaciones puntuales o variaciones en el número de copias en SCN1A en el 80% de los casos (de novo en >95%). La epilepsia asociada a mutaciones en SCN1A es un buen ejemplo de heterogeneidad feno típica de las epilepsias genéticas. Aunque son la causa más frecuente del síndrome de Dravet, se han identifi cado mutaciones en SCN1A en pacientes con fenotipos benignos (convulsiones febriles, GEFS+, síndrome de Panayiotopoulos) o de tan mal pronóstico como la epilep sia migratoria, el síndrome de West o en la encefalopatía epiléptica precoz8.
Los fármacos más utilizados en el tratamiento incluyen ácido valproico, clobazam, topiramato y stiripentol. La dieta cetogénica, levetiracetam, zonisamida, perampanel, etosuximida pueden ser también de utilidad. Stiripentol, cannabidiol y fenfluramina son los únicos que disponen de un ensayo clínico para esta indicación.
Los bloqueantes del canal de sodio como lamotrigina, fenitoina o carbamazepina pueden determinar un agrava miento de crisis y están contraindicados.
Se describen mutaciones en otros genes responsables de cuadros similares (referidos a veces como síndromes de Dravet-like) entre los que destaca la protocadherina 19 (PCDH19), localizada en el cromosoma X y responsable del ‘síndrome de retraso mental (con o sin epilepsia) limi tado a mujeres”. Otros genes identificados con el fenotipo de Dravet son GABRA1, STXBP1, CHD2, SCN1B, SCN2A y raramente KCNA2, HCN1, and GABRG218,19.
Los avances en los últimos años en reconocimiento clínico, electroencefalográfico, estudios moleculares y trastornos metabólicos asociados a las encefalopatías epilépticas han permitido mejorar el enfoque terapéuti co y el pronóstico de los pacientes con encefalopatías epilépticas intentando una medicina de precisión. La unificación de criterios diagnósticos para cada síndrome sugerida recientemente por la ILAE facilita su identifica ción y el manejo temprano de los mismos puede cambiar la evolución de muchos pacientes.

