











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Allergic rhinitis (AR) is a disease that affects around 40% of the world population, whereas Mexico reports an estimate of total prevalence of 15%.1,2 The two most common symptoms that most strongly affect the patient’s quality of life are rhinorrhea and nasal congestion. Half of the pa tients with AR in Mexico have persistent rhinitis, and the congestive component is present in almost 90% of the patients.3
AR can be classified as persistent when the symp toms occur 4 or more days a week or during 4 or more weeks.4 Moderate to severe symptoms affect the patient’s capacity to do daily activities and are associated with fatigue, changes in the patient’s mood, cognitive disorders, depression and anxiety.5 AR treatment requires preventive measures such as avoiding contact with the allergen as much as possible or, the most common treatment, phar macotherapy.6 In that sense, the ARIA (Allergic Rhinitis and its Impact on Asthma) Guidelines recommend the use of intranasal corticosteroids and second-generation anti-histamines as first-line treatment, as well as the use of anti-leukotrienes or immunotherapy, when Persistent Allergic Rhinitis PAR doesn’t respond to primary treatment.4,7,8
Montelukast is an anti-leukotriene that binds with high affinity and selectivity to the cysteinyl-leukotriene receptor 1 (CysLTR-1), thus inhibiting the physiological actions of leukotrienes C4, D4 and E4, directly associated with the symptoms of AR.9,10 Desloratadine, on the other hand, is a second-generation antihistamine, selective antago nist of histamine H1 receptors. It doesn’t penetrate the central nervous system and has high affinity for such receptor compared to cetirizine, ebastine, loratadine and fexofenadine; in addition, desloratadine has a longer half-life (27 h), which produces a substantial benefit in nasal and ocular symptoms in patients with moderate AR as opposed to other second-generation antihistamines.11-13
The combination of these two drugs is a com prehensive treatment for the allergic process; it is aimed at different molecular targets within the physiopathological process of PAR. The thera peutic effects of desloratadine theoretically have advantages over loratadine, since it is considered the active metabolite of the drug. Also, given the fact that this is a convenient treatment (only 1 time a day), it can contribute to patient compliance and successful pharmacotherapy.
Even though there is evidence on the efficacy and safety of this combination for PAR14,15,22,23, it is not available in the Mexican market, so it is necessary to show the efficacy and safety prior to requesting sanitary registration from the regula tory authority. So, the objective of this study was to evaluate the efficacy and safety of the fixed-dose combination of montelukast/desloratadine 10 mg/5 mg in comparison with montelukast/ loratadine 10 mg/10 mg administered once a day for 6 weeks.
MATERIALS AND METHODS
Study design and population
Controlled, randomized, double-blind, therapeutic confir matory, prospective, longitudinal, parallel-group, multicen ter clinical trial including Mexican adult patients diagnosed with PAR of at least one year of evolution with moderate to severe signs and symptoms according to the ARIA classification, and a baseline SNOT-20 score of at least 3 points. Exclusion criteria: patients with history of asthma, hypersensitivity to any of the study drugs or formulation excipients, recent respiratory infections, history of rhino sinusitis, problems with nasal structures, including nasal polyps, septum deviation (around 70%) that significantly impact the nasal airflow, patients addicted to steroids or decongestant inhalers, pregnant or lactating women, use of acetylsalicylic acid or concomitant use of immunotherapy or antihistamines that couldn’t complete an elimination lavage period of at least 7 half-lives before enrollment.
Patients were enrolled after they signed their informed consent. The protocol and every document that has been delivered or applied to patients were previously approved by Research Ethics Committees and Research Committees in accordance with the local rules. All the procedures were performed in accordance with the Declaration of Helsinki and Good Clinical Practice (ICH E6R2).
Research centers were distributed in different states of the Mexican Republic, including the Otolaryngology Service of the Hospital Médica Sur (City of Mexico, Mexico), the Instituto de Investigaciones Aplicadas a la Neurociencia, A. C. (Durango, Mexico), and Ícaro Investigaciones en Medicina, S.A. de C.V. (Chihuahua, Mexico).
Treatments
Research subjects were randomized at a 1:1 ratio to the treatment arm with the study drug montelukast/desloratadine 10 mg/5 mg capsules or to the comparator arm with montelukast/loratadine 10 mg/10 mg tablets (Montaclar®), both treatments administered every 24 hours (evening dose) orally for 6 weeks.
Study variables
Treatment efficacy was determined through the global score of the SNOT-20 (Sino-Nasal Outcome Test) questionnai re,16 and also through information collected from medical history, physical exploration with previous rhinoscopy, and the scores of the T5SS (Total 5-Symptom Score),17 and TSQM (Treatment Satisfaction Questionnaire for Medica tion) questionnaires.18 The tests were conducted during ba seline assessment (day -7), at the start of treatment (day 1), on day 21 (follow-up) and at the end of treatment (day 42).
The primary efficacy variable was established as the difference between the baseline global score of the SNOT-20 questionnaire and the global score obtained in week 6. If the difference between the baseline and final score is more than zero (positive), it is interpreted as a favorable result, but if the value is less than zero, it is interpreted as an unfavorable result. A change of more than 3 points in the global score of SNOT-20 was considered an improvement of clinical relevance.
Secondary efficacy variables are the area under the curve (AUC) of the SNOT-20 of each visit, SNOT-20 indicators per treatment and per visit, the severity classification of SNOT-20 scores, the T5SS questionnaire, T5SS indicators per treatment and per visit (21 days and 42 days), severity classification of T5SS scores, use of the rescue drug (inha led mometasone, prohibited during the first 10 days, use allowed for 2 weeks, maximum), and also the scores of the TSQM questionnaire.
Statistical analysis
Sample size was calculated taking into account the standard deviation (SD) of 1 point in the SNOT-20 questionnaire reported by Piccirillo et al, 200219 and a delta of 0.8, which is considered clinically significant by the same author. The sample was calculated with the PASS 13 program, also considering a significance level of 2.5% and 90% power for the non-inferiority hypothesis. Taking into account 20% of withdrawals, the sample was established in 86 patients.
The statistical analysis used the Student’s t Test or the Mann-Whitney U Test for the comparison of mean values. For the analysis of the variables on a categorical scale (nominal or ordinal), the Fisher’s Exact Test was used. Demographic and baseline characteristics are presented with descriptive statis tics. The statistical analysis was carried out using the Stata® 15 (StataCorp, Texas, United States), NCSS® 11 (NCSS, LLC. Kaysville, Utah, United States) and East® version 6 (Cytel Inc, United States) programs. The significance level for variable analysis was set at 5% (Type I error, α = 0.05), except for the non-inferiority test, whose level of significance was set at 2.5% (Type I error, α = 0.025) for being unilateral.
RESULTS
In the present study, 44 patients were enrolled for the group that received treatment with the active comparator, MKLOR (montelukast/loratadine), and 42 patients were included in the test group, MKDES (montelukast/desloratadine), for a total of 86 patients randomized for the intention-to-treat (ITT) population. During the database review, under double-blind conditions, subjects with baseline SNOT-20 scores < 3 (visits on day -7 and day 1) were discarded, according to the eligibility criteria, leaving 37 patients in each group for a total of 74 individuals in the per protocol popula tion (Figure 1).

Figure 1 CONSORT flow diagram. It reflects the process of patient selection. randomization and analysis. Per protocol population n=74. ITT: intention-to-treat.
The per protocol population (n=74) allowed the evaluation of efficacy variables (primary and secondary), whereas in the intention-to-treat population (n=86), the demographic and clinical variables, the TSQM questionnaire, and safety and tolerability variables were evaluated for the con firmatory analysis of the primary efficacy variable.
62.8% (n=54) of the 86 patients were female, however, the demographic variables and data from medical records (vital signs) didn’t show clinically relevant differences between treatment groups (Table 1).

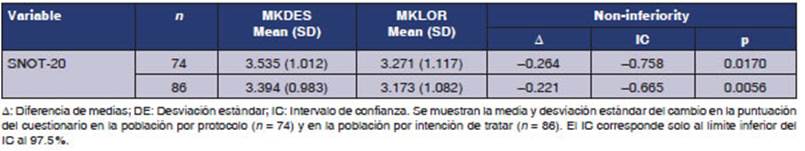
The primary efficacy analysis showed that the change in the global score of the SNOT-20 ques tionnaire was more than 3 points for both groups in the per protocol population, with a value of 3.54 points in those treated with MKLOR (-0.78 to 4.80) and 3.27 points (0.03 to 4.35) for MKDES; the difference in means (test-reference) was -0.26 points, with a 97.5% CI lower limit of -0.76 points, not exceeding the clinically relevant inferiority margin of -0.8 (p=0.0170).19 Therefore, treatment with MKDES is not inferior to MKLOR in terms of efficacy for the treatment of PAR symptoms. This was verified by the ITT population (p=0.0056), with a difference of means of -0,22 points and a 97,5% CI lower limit of -0,67 points (Table 2).
The potential impact of the demographic vari ables was evaluated by linear regression, consider ing the change in the global SNOT-20 score as the dependent variable, and the treatment, research site, age, gender, and body mass index as inde pendent variables. Only the research site had a significant effect on the primary efficacy variable (p <0.0001). Individuals from one center had a smaller change in score compared to the other centers. Since this occurred in only 10 patients, it was not considered to have a significant impact on the conclusion of the non-inferiority test, and was considered a common finding of multicenter studies.
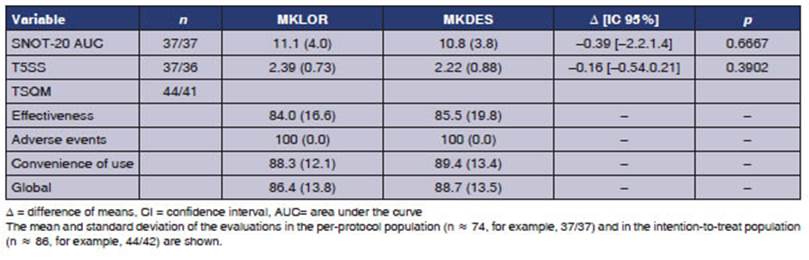
In the secondary efficacy analysis, global scores from the baseline measurement to the last week showed a mean difference of -0.393 units of area under the curve, with no significant difference between the groups (p = 0.6667). No significant difference was found for the global change of the T5SS (p = 0.3902), which is consistent with the conclusion of on-inferiority of the primary efficacy variable. The global TSQM score was greater than 80% in both groups, as were the dimensions of effectiveness and convenience of use; the adverse events dimension suggested a high degree of toler ability (Table 3).
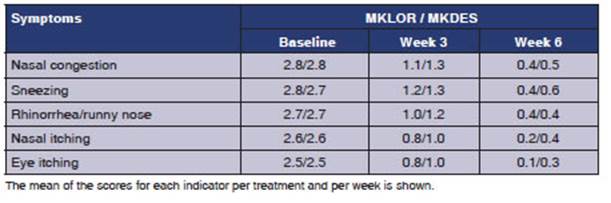
The indicators of the SNOT-20 questionnaire were evaluated by group and by week; both treat ments reduced the scores of the indicators with no differences between the indicators of symptoms or quality of life (Table 4). The T5SS indicators evaluated by group and by week showed that both treatments reduced the score, with no differences between groups (Table 4).

The severity levels of SNOT-20 indicators in baseline conditions classified about 75% of patients in the following levels: “4 severe” or “5 can’t be worse” for both groups. At the sixth week of treat ment, more than 90% (91.9% MKLOR and 91.7% MKDES) of patients were classified as “1 very mild” or “0 no problem”. The severity levels of T5SS indicators in baseline conditions classified more than 70% of patients in maximum level, “3 severe” for both groups. At the sixth week of treatment, more than 90% (91.9% MKLOR and 91.7% MKDES) of patients were classified in the following levels: “1 mild” or “0 none”, indicating that both treatments globally improved the five symptoms under evaluation (Table 5).
The use of the rescue drug occurred only in 5 of the 86 intention-to-treat patients. Four patients of the MKDES group were recruited in the same center. 2 of the 5 patients used the rescue drug in the third week; 3 patients used it during the sixth week; and the amount of time they used said drug varied from 1 to 18 days. The small number of patients who used rescue medication does not allow us to infer whether its use and duration were related to the result of the variables in the different treatment groups.
Adverse events (AEs) occurred in 4 of the 86 patients. A total of 12 AEs were reported. Three patients from the MKLOR group reported 8 AEs and one patient from the MKDES group reported 4 AEs. One patient showed elevated aminotrans ferases (> 2 times the reference value), without concomitant medication, and the investigator attributed it to the MKLOR drug. The event was solved after the subject suspended treatment and showed an improvement. None of the treatment groups showed serious adverse events.
DISCUSSION
For some time, leukotriene receptor antagonists (LRAs) were considered secondary treatment for PAR in patients with asthma. The information available for PAR without asthma in the 2010 ARIA review showed LRAs with a small benefit in preschool children, limited efficacy in adults, and a high cost, therefore the recommendations pointed towards oral antihistamines with a clinical value that was higher than LRAs.20 A large number of patients with AR don’t go to the medical consulta tion because they believe that their symptoms are “normal”; others use over-the-counter medication, and only a small part go to consultation where they are diagnosed with moderate or severe PAR.21 In the present study, the profile of selected patients had a minimum of 3 points in the SNOT-20 score. These patients could benefit from a combination with the suitable power and sustained action.
The combined use of antihistamines and an tileukotrienes has been reported to have advan tages in terms of efficacy over monotherapy in patients with PAR. For example, the combination of montelukast and desloratadine or levocetiri zine decreased nasal symptoms and the levels of eosinophil cationic protein above what had been observed for the drugs alone.22 The advantage of the therapeutic combination in terms of health-related quality of life and the nocturnal symp toms scale, obtained from the Rhinoconjunctivitis Quality of Life Questionnaire (RQLQ) has also been verified; in addition, the presence of adverse events was similar for placebo, montelukast, levo cetirizine or the combination of montelukast and antihistamines.23
In the present study, treatment with monte lukast plus loratadine or desloratadine achieved a difference of more than 3 global points in the SNOT-20 questionnaire in patients with PAR on week six; this change is clinically relevant and shows the therapeutic utility of the combination. The 0.8 delta in the SNOT-20 score is considered clinically significant,16,19 thus, the difference in means reported here with a confidence interval within a margin lower than said cut-off point al lows us to affirm that the MKDES study treatment is not inferior to the MKLOR active comparator. The follow-up time used in this study was compa rable to previous studies evaluating the clinical effects of treatment with montelukast and antihistamines, 22 though shorter than others;23 however, the design of the present study and the SNOT-20 and T5SS instruments have demonstrated clini cally relevant changes with adequate coverage of the proposed objective, both in the per protocol population and in the intention-to-treat popula tion. The evaluation time of six weeks of treat ment and total evaluation are justified according to the criteria of other authors;24 furthermore, the evaluation period is appropriate for the SNOT-20 primary efficacy instrument in accordance with the validation history,16,19 as well as the TSQM questionnaire. Reported TSQM scores of 84% to 100% are indicative of high patient satisfaction and excellent tolerability to the combined treatment.
A limitation of this study is that the evaluation window does not allow checking how symptoms be have with a long-term treatment, for example, the XPERT study for PAR treated with levocetirizine evaluated nasal and ocular symptoms with T5SS from 4 weeks to 6 months of treatment in order to report the moment in which the symptoms started to improve and which remained stable throughout the whole treatment. In this study, levocetirizine improved nasal congestion significantly after the first month of treatment and continued that way for more than 6 months.18 The evaluation window here was sufficient to verify that the symptoms improved from week 3 with indicators in the mean score of “1 mild”, and in week 6 with the mean score close to “0 none”. The evaluation of Ciebiada et al for 32 weeks demonstrated the long-term effect of montelukast in combination with desloratadine or levocetirizine. However, their evaluation used a different instrument focused on nocturnal symptoms.23 Future studies could evaluate the long-term effect of the combination of antileukotriene plus antihistamine in PAR in the Mexican population.
The severity levels of the indicators in the SNOT-20 and T5SS questionnaires showed that quality of life conditions and symptoms had very high scores in the baseline evaluation: 7 to 8 of every 10 patients were in the highest levels of severity; and six weeks after the beginning of the study, the scores decreased to a minimum in 9 of every 10 patients; this improvement was achieved in both treatment groups.
Expected AEs observed in patients receiving the treatments under evaluation were: cephalea, dyspepsia and gastrointestinal discomfort, related to the use of montelukast or desloratadine.24,25 Loratadine doesn’t seem to be related to these symptoms; however, we observed these AEs in both treatment groups with clinical characteris tics that did not show a clear causal relationship. One patient from the MKDES group presented AEs described as “gastritis”, “colitis” and “diar rhea”, whereas a patient from the MKLOR group had “acute gastritis”. In both cases the causal ity was reported as “unclassifiable”. Cephalea occurred in a patient from the MKLOR group, with causality reported as “conditional” due to concomitant consumption of alcohol, even though it was prohibited. In most cases, concomitant medications were used to resolve the manifesta tions. As reported in the literature, the combi nation of the drugs under evaluation was safe, considering that no serious was reported during the study; all reported AEs were milder moderate, only one of them was from the MKDES group, and one drug-related AE in the MKLOR group was self-limiting at the end of treatment. With that being said, it is possible to conclude that the montelukast/desloratadine study medication had adequate tolerability.
CONCLUSIONS
The combination of desloratadine and montelukast significantly improves symptoms in patients di agnosed with PAR. Treatment evaluation showed clinically relevant efficacy and safety that weren’t inferior to the combination of montelukast and loratadine. These results suggest that the oral com bination of montelukast + desloratadine 10 mg/5 mg is a good treatment option for adult patients who require a drug with an action mechanism dif ferent from that of antihistamines for controlling the signs and symptoms of the disease.

