











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkSignal transducer and activator of transcription 3 (STAT3) is a protein encoded by the STAT3 gene that con trols cell proliferation, growth and apoptosis through the regulation of gene expression in response to extracellular cytokines, hormones and growth factors1. The germline gain of function of the STAT3 gene causes an autosomal dominant disease, clinically characterized by a significant lymphoproliferation, including lymphadenopathy and/or hepatosplenomegaly, as well as childhood onset autoim munity1. Autoimmune cytopenias are common, including autoimmune haemolytic anaemia, neutropenia and/or idiopathic thrombocytopenia. Patients may also show eczema, hypogammaglobulinemia, severe and recurrent infections, and growth failure1-3.
Common variable immunodeficiency (CVID) is the most frequent symptomatic antibody deficiency, characterized by an impaired antibody production. Over 90% of patients suffer from extracellular capsulated bacterial infections, mainly of the respiratory tract; 25-30% develop autoim mune diseases, and 8-22% benign lymphoproliferative or granulomatous disease. These patients are also more susceptible to oncological diseases, mainly lymphomas and gastric adenocarcinoma. The most common auto immune diseases are idiopathic thrombocytopenia and autoimmune haemolytic anaemia. Lungs, lymph nodes and spleen are the most frequently affected organs due to benign lymphoproliferative disease4.
We present an adult patient who, during his first years of life, presented severe benign lymphoproliferative dis ease, recurrent infections and autoimmune haemolytic anaemia and was initially diagnosed as CVID. During his adulthood and after a diagnostic review, a gain-of-function (GOF) mutation in STAT3 was found. STAT3 GOF was first described about 35 years after our patient disease onset. When molecular diagnosis could be made, he al ready had significant deterioration of lung function, limiting the therapeutic possibilities.
Clinical case
The patient was a 40-year-old male; son of non-consanguin eous and healthy parents. He developed lymphadenopathies at 18 months of age. A lymph node biopsy was performed, reporting non-specific lymphoid hyperplasia with a preserved follicular structure. At 21 months, five and seven years of age, he presented lymphadenopathy again with the same results in lymph node biopsies. When he was five he developed hepatoesplenomegaly. He had several episodes of pneumonia during childhood, with good response to antibiotic treatment. At the age of eight, autoimmune haemolytic anaemia was diagnosed and treated with corticosteroids evolving with ex acerbation and remission periods. When he was 13 years old, he was referred to a pediatric immunologic center where low serum IgG, IgM and IgA levels were found, with an impaired post-vaccination antibodies response, low hemagglutinin titers but normal number of B lymphocytes. CVID was diagnosed and he began treatment with intravenous immunoglobulin replacement.
At the age of 20, he had a new episode of pneumonia, where a thorax computed axial tomography showed mediasti nal adenopathies, pulmonary infiltrates adopting ground glass configuration, and accentuation of the reticular interstitium with interlobular thickening diffusely compromising both lung fields (Fig. 1). A lymph node and a pulmonary biopsy were performed, reporting nonspecific lymphoid hyperplasia and lymphocytic interstitial pneumonia, respectively, and treatment with corticoids was performed with good response. At the age of 22, he started follow-up in our hospital unit. Enterovirus meningoencephalitis was diagnosed and was treated with acy clovir until HSV was ruled out and the interval between doses of immunoglobulin was reduced, receiving one dose upon hospital admission. Infection resolved with residual seizures. He had lymphocytic interstitial pneumonia relapse when turn ing 24 years old and was treated with steroids at high doses. A lymph node biopsy performed 10 years afterwards reported lymph node hyperplasia. Several lymphoid phenotypes showed lymphopenia, inversion of CD4/CD8 ratio, normal B cell count, CD21low expansion, decreased switched memory B cells, normal double negative (CD3+ TCRαβ + CD4- CD8-) and NK cells. An autoimmune lymphoproliferative syndrome (ALPS) was excluded based on a normal double negative cell count and normal levels of vitamin B12 and soluble FasL expression.
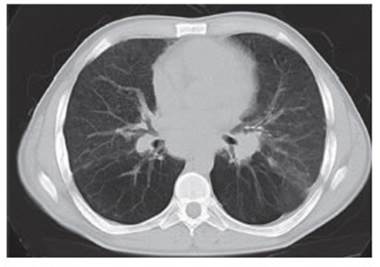
Fig. 1 Thorax computed axial tomography. Pulmonary infil trates adopting ground glass configuration, accentuation of the reticular interstitium with interlobular thickening diffusely compromising both lung fields
Considering the association of respiratory infections, auto immunity and benign lymphoproliferation of childhood onset, genetic studies were decided. In our country it is still difficult to access sequencing techniques, which, in many cases, delays and makes diagnosis difficult. Patients without health insurance cannot access these type of studies. After signing informed consent, the patient was enrolled in a National Insti tutes of Health (NIH, Bethesda, MD) diagnostic protocol (05- I-0213 Screening and Baseline Assessment of Patients with Abnormalities of Immune Function, registered at ClinicalTrials. gov) and whole exome sequencing (WES) was performed according to the manufacturer’s protocol (Illumina Exome with Enrichment Flex/HiSeq 2500). A heterozygous mutation, STAT3 NM_003150 c.2141C>T, p.P714L was detected by WES and confirmed by Sanger sequencing (Life Technologies, BigDye Terminator v1.1 Cycle Sequencing Kit/3500 Genetic Analyzer) (Fig. 2). Previously published functional studies showed that this mutation resulted in GOF5.
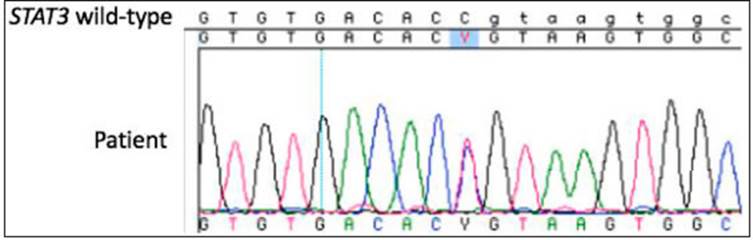
Fig. 2 Sanger sequencing confirmation of WES-detected heterozygous variant STAT3 NM_003150 c.2141C>T, p.P714L. The patient’s Sanger sequencing trace data was aligned to the STAT3 NM_003150 reference (wild-type) sequence. The IUPAC-IUB code ‘Y’ in the patient’s data denotes the presence of both cytosine (C) and a thymine (T) peaks at that position
During the last years the patient developed multiple respi ratory infections, progressive decline in lung function without significant changes in thorax computed tomography scans, and right ventricular systolic function impaired. He died at the age of 41 due to an infectious complication that worsened his lung disease. He was waiting for a lung transplant.
Discussion
STAT3 GOF germline mutations were first described in five individuals with childhood-onset autoimmunity by Fla nagan et al. in 20141. Shortly thereafter Haapaniemi et al. reported three patients with de novo mutations, clinically characterized by multiorgan autoimmunity, lymphoprolif eration, and late-onset mycobacterial disease2. Milner et al reported nine heterozygous germline mutations in thirteen individuals from ten families with lymphoproliferation au toimmunity and early-onset organ autoimmunity3. A total of 43 cases were later reported by different authors5,6.
STAT3 GOF disease has an autosomal dominant inheritance pattern. Although most identified mutations were de novo, six members of five families carrying a STAT3 mutation were asymptomatic or had a less severe phenotype, indicating that there were carriers who showed an autosomal dominant inheritance with incomplete pen etrance3. Since the parents of our patient did not show symptoms of the disease, but they were not studied, this defect could correspond to a de novo mutation but it can not be ruled out that it is an autosomal dominant mutation with incomplete penetrance.
In most patients, genomic sequencing was necessary because the diagnostic approach is challenging, since clinical manifestations are variable and the similarity with other lymphoproliferative syndromes makes its presump tion difficult. Initially, when STAT3 GOF disease had not been yet described, a diagnosis of common variable im munodeficiency was made based on his clinical history of infections, autoimmune cytopenias, hypogammaglobu linemia and decrease switched memory B cells.
Lymphocyte phenotype as a diagnostic tool in STAT3 GOF disease is controversial since the findings are vari able, as are the levels of immunoglobulins4-6. Lympho proliferation was accompanied by an increase in CD3+ CD8- CD4- double negative T-cell count and defective mediated Fas apoptosis in some series3,7. An overlap in the phenotype with ALPS suggests that germline GOF mutations in STAT3 produce a strong susceptibility to immune dysregulation. According to Nabhani et al, Fas expression was decreased and Fasmediated apoptosis was deficient in patients with STAT3 GOF mutations and ALPS like phenotype7. Thus, these diseases may have a common pathophysiologic mechanism at some point, involving the failure of self-tolerance. Consistent with this, our patient has an ALPS like phenotype. It is interesting to note that our patient’s mutation had previously been described in two siblings diagnosed with ALPS, in whom clinical evolution led to the suspicion that there was an other underlying mutation5.
Therapeutic decisions are difficult to make due of a lack of prospective studies and the scarce number of reports. Immunoglobulin replacement therapy is indicated in patients with hypogammaglobulinemia and recurrent infections. It has been reported that the use of immunosuppressants like corticosteroids, mTOR inhibitors, mycophenolate mofetil and rituximab is the mainstay of the treatment for autoim mune manifestations6-11. However, the use of immunosup pressants could have significant side effects and often doesn´t result in clinical resolution of lymphoproliferative and autoimmune manifestations. Eight patients were suc cessfully treated with tocilizumab, a monoclonal antibody against the Il-6 receptor, and specific inhibition of JAK molecules, including tofacitinib and ruxolitinib3,8-11. Until now, hematopoietic cell transplantation was performed in five patients, four of whom died3,10. This would suggest that other therapeutic approaches, such as specific small-molecule inhibitors, are perhaps more appropriate therapies.
STAT3 gain of function disease was described about 35 years after our patient disease onset. When molecu lar diagnosis could be made, he already had significant deterioration of lung function. New promising treatments such as JAK inhibitors weren´t available in our country at that time.
The diagnosis of STAT3 GOF mutations should be suspected in patients with lymphoproliferative disease, autoimmunity and hypogammaglobulinemia. It is important to consider this diagnosis in CVID patients that share these characteristics, regardless of their age. In order to deter mine the therapeutic efficacy of JAK inhibitors, Il-6 receptor antagonists and hematopoietic cell transplantation, long-term studies are needed. Nowadays, early diagnosis may allow the implementation of targeted precision therapies that could modify the disease evolution.

