











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkKEY POINTS
• Population-based epidemiological inves tigations are essential for comprehend ing and quantifying the repercussions of uncommon diseases. These studies aid in heightening awareness, improving diag nosis, and guaranteeing optimal care and assistance for those impacted by HHT. No tably, there is a deficiency in the scientific literature concerning HHT prevalence, spe cifically in Latin America.
• The prevalence of HHT identified in our study is higher than the one documented in numerous studies. This is the first preva lence report in the Latin American popula tion.
Hereditary hemorrhagic telangiectasia (HHT) also known as Osler-Weber-Rendu syndrome, is an autosomal dominant vascular dysplasia that might affect 1/5000-10 000 individuals world wide1. Is a rare condition and the underdiagno sis is frequently described2. It is mainly caused by heterozygous mutations of the endoglin gene (ENG) HHT type 1, activin-like receptor kinase 1 (ACVRL1) HHT type 2, and MADH4 (SMAD4) leading to overlap syndrome with Juvenile Pol yposis. All genes belong to the BMP/TGFβ signal ing pathway3,4.
A definitive diagnosis is based on the pres ence of at least three Curaçao criteria: recur rent epistaxis, mucocutaneous telangiectasias, arteriovenous malformations (AVMs) in typical internal organs (brain, lung, liver, gastrointesti nal tract), and a first-degree relative according to these criteria and/or a positive genetic test5.
HHT is characterized by mucocutaneous tel angiectasias localized mainly in lips, face, hands, and tongue, and arteriovenous malformations (AVMs) in organs such as the central nervous system, lung, liver, and the digestive tract. The most prevalent symptom is the epistaxis (95% of cases), typically spontaneous and recurrent and can range from mild to life-threatening affecting the quality of life and leading to iron deficiency anemia6. Other symptoms are related to the or gan affected. Brain hemorrhage or seizures may occur due to brain vascular malformations. Pul monary AVMs may cause ischemic stroke, brain abscesses, hypoxemia, or hemoptysis. Hepatic vascular malformations are usually asymptom atic, although some patients suffer from high output cardiac failure, portal hypertension of biliary tract necrosis7,8.
HHT exhibits age-related penetrance, and the average age at which telangiectasias and AVMs develop and manifest symptoms varies signifi cantly depending on the specific organ involved9. An average time lag of 25 years separates dis ease onset and the first definite diagnosis10. Lack of knowledge and awareness about HHT clinical features among health workers can significantly delay the diagnosis increasing morbidity. Proper and early screening methods are necessary to avoid clinical complications1.
Population-based epidemiological studies play a crucial role as the initial step in comprehend ing and quantifying the impact of this disease. By examining the burden of disease, such studies help inform healthcare planning and resource al location for a condition that is often under-rec ognized. They contribute to raising awareness, improving diagnosis rates, and ensuring appro priate care and support for individuals affected by HHT.
Currently, there is a shortage of scientific lit erature regarding the prevalence of HHT, and specifically, there is a notable absence of such reports in the Latin American region. We aim to estimate the prevalence in the Hospital Italiano Medical Care Program (IHMCP), a prepaid health maintenance organization (HMO) in Buenos Ai res, Argentina.
Materials and methods
A descriptive cross-sectional study was designed, which included all patients over 18 years of age affiliated at the IHMCP.
IHMCP provides comprehensive medical and health services currently with over 172 514 affiliates. Our center has been a tertiary-level university hospital and an HHT Reference Center since 2010, holding an Institutional Reg istry of Hereditary Hemorrhagic Telangiectasia (Clinical trials.gov NCT01761981) and receiving patients’ consulta tions from different cities and neighboring countries.
In order to meet the criteria for case inclusion, indi viduals were required to be aged 18 or older, have an af filiation with the IHMCP, and possess a clinical diagnosis of HHT based on the Curaçao criteria and/or a positive genetic test result. Case detection included the search in our Institutional Registry of HHT in the RedCap electronic platform (Research Electronic Data Capture). Data collec tion was obtained from the medical records of each pa tient included in our Institutional Registry.
For the patient description, descriptive statistics were employed. Categorical variables were reported as relative frequencies and/or percentages, while quantitative vari ables were expressed as mean and standard deviation (SD), or median and interquartile range (IQR), depending on their distribution.
The prevalence was calculated by dividing the number of cases by the total number of active affiliates at January 2023 (151 035).
Also, age and gender specific prevalence rates were es timated. Rates are expressed by 10 000 people with their 95% confidence intervals (95% CI).
Rates were standardized by the age and gender dis tribution of Europe, the USA, and the global population according to the Argentinian 2010 census.
Descriptive statistical analysis was performed using the Stata 16 version.
The project obtained approval from the institutional ethics committee. Information was stored confidentially and restricted to the researchers.
Results
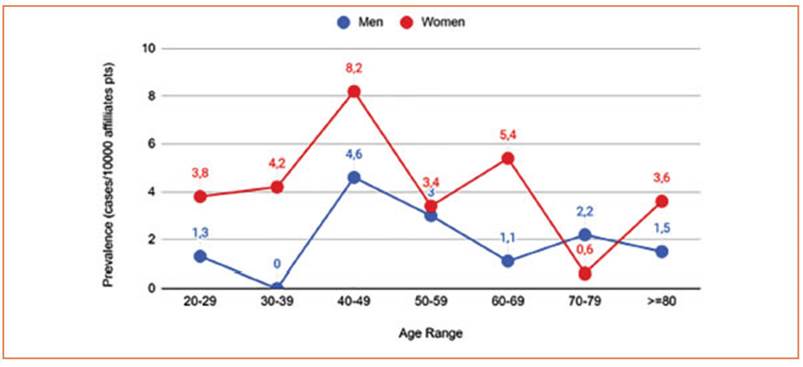
This study reported 48 HHT cases according to fulfilled Curaçao criteria and/or positive ge netic diagnosis. We estimate the prevalence as 3.2 in 10 000 (IC 95% 2.4-4.2). Specific prevalence in women was 3.9 in 10 000 (IC 95% 2.8-5.5) and in men 2.1 in 10 000 (IC 95% 1.2-3.6) (Table 1). The prevalence of HHT diagnosis showed a notewor thy association with sex, with a higher occur rence in women compared to men. Out of the 48 patients, 35 were women (72.9%) with an aver age age of 55 (19.9), and the average age for men was 55 (17.2). The average age of the 48 patients was 55 (SD 19). Age rates by gender were calcu lated (Fig. 1).

Table 1 Hereditary hemorrhagic telangiectasia (HHT), prevalence estimated by sex. In Hospital Italiano Medical Care Program (IHMCP)
In relation to patient referral sources, the most prevalent was a referral by another physi cian (60.4%), followed by family history (18.7%). Other sources included the institution’s journal, information on the internet, or from other pa tients. The 48 patients corresponded to 39 fami lies.
Regarding the age and sex-standardized prev alence to the worldwide population, the preva lence rate is 2.03 (IC95% 1.35-2.7). In addition, the age and sex-standardized prevalence for the USA population is 2.43 (IC95% 1.67-3.18) and 2.40 (IC95% 1.65 - 3.15) for Europe.
Discussion
This report estimated a prevalence of HHT of 3.2 in 10 000 patients (IC 95% 2.4-4.2) showing that HHT is a rare health problem, consistent with the findings of many worldwide reports. There is no local-level information available on the epidemiology of HHT, and there are few in ternational reports.
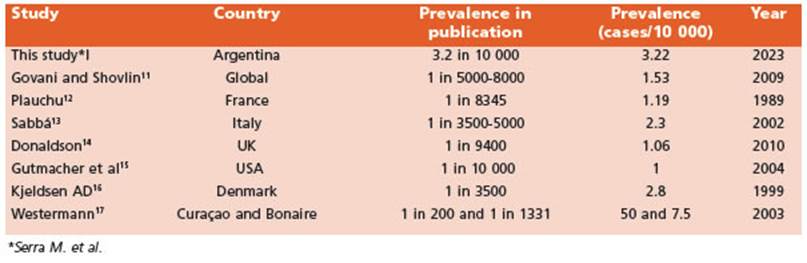
Our age and sex-standardized prevalence, when applied to the global population, is 2.03/10 000. According to the most recent and cited reviews, the estimated worldwide preva lence is 1:5000-8000 people11. However, it is worth noting that this review is primarily based on two articles from isolated populations in France and Denmark, which have a high concentration, probably due to the “founder effect”, making it difficult to compare to our population.
The most detailed study on the prevalence of HHT was presented in 1989 by Plauchu et al. as a result of research conducted on the French pop ulation, studying different departments of the country through a questionnaire sent to clinical professionals. They estimated a prevalence of 1 in 834512. However, the methods applied were different from the ones used in our research. Sabbá et al. reported in 1/3500-5000 in 2002 in Italy13 and the most recent work, dated 2010 in the UK, estimated a prevalence of 1 in 9400 in dividuals14. They used The Health Improvement Network, a longitudinal, computerized general practice database covering 5% of the UK popula tion to identify all recorded diagnoses of HHT. These results are similar to the age and sex-standardized prevalence of 2.4/10 000, using de European population as standard population. In addition, we found an age and sex standardized prevalence of 2.43/10 000 to the USA population, similar to the Guttmacher study, that reported 1 in 10 000 through a genetic epidemiology study in 200415.
Denmark, specifically the County of Fyn (one of the main islands of Denmark) has a preva lence of 1/3500 (1999), which is quite similar to ours. They conducted a study based on two cross-sectional surveys combined with a long-term fol low-up study16. Their prevalence is relatively high compared to other studies. They suggest that this difference may be due to variations in study de signs, possibly combined with geographical dif ferences in the distribution of HHT.
In certain regions such as Curaçao and Bo naire, notably high prevalence rates were docu mented (1 in 200 and 1 in 1331, respectively)17,18. Thirteen families of Antillean descent were in volved. A total of 219 members were examined, 51% of patients have definite HHT diagnosis, reaching the point-prevalence of 1 in 1331 inhabitants. Nevertheless, it was not feasible for us to standardize our rate to these particular populations. However, it is crucial to acknowl edge that this elevated prevalence could be attributed to the “founder effect”, resulting in re duced genetic diversity within the population.
For visual representation, we standardized the calculation of all previously cited prevalence values by employing a uniform denominator of 10 000. This is illustrated in Table 2.
In addition, same as other studies2,17, we also found a higher prevalence in women. Due to the autosomal dominant nature of HHT, there is a 50% likelihood for the offspring of affected individu als to inherit the mutation, and it is important to note that this inheritance does not inherently exhibit a gender bias. The phenomenon reported might occur due to females’ higher attendance at the health system and might reflect a higher misdiagnosis in the male gender. HHT manifes tations, particularly nose and gastrointestinal bleeding, tend to worsen as patients age or dur ing pregnancy19. This may also help explain the higher prevalence of HHT diagnosis in females. Further research is crucial to clarify whether the difference in HHT diagnosis between sexes is solely attributed to behavioral factors or if there might be biological reasons involved as well.
Our research aligns with existing studies that have also observed a higher prevalence of HHT in adults14. Our findings suggest that the disease may have age-related penetrance or that the like lihood of diagnosis is influenced by longer lifes pans. This is why the median age of these patients may indicate a delay between the onset of symp toms and diagnosis time. It may also reflect the lack of knowledge of HHT among health work ers and members of the affected families. In this setting, prevalence studies may help to highlight rare diseases to be considered by the health work ers in their daily professional practice. Moreover, it should be emphasized that patients under 18 years of age were not included in this study. Even more, HHT Clinical Guidelines establish that a ge neticist test must be performed in neonates born to parents with a history of the condition1.
Additionally, it is often observed that individ uals within the same family, who have a high likelihood of being diagnosed with HHT based on the presence of typical symptoms, either re frain from seeking medical diagnosis or deny specific information. This situation may be at tributed to outdated and incorrect beliefs held by both patients and some physicians regarding the availability of curative treatments for HHT. On the other hand, late diagnosis could increase the risk of adverse consequences, morbidity events, and even sudden death at any age. Be ing this a disease with an impact on multiple systems and organs, its approach is multidisci plinary and complex in order to follow up and care for patients and their families. Additionally, the necessity for a prompt diagnosis can be em phasized not only in terms of improving quality of life but also in reducing the financial burden10.
Currently, one of the primary obstacles in prevalence studies is the presence of undiag nosed patients, which can be attributed to the medical community’s limited understanding of HHT as well as other rare diseases. These con ditions have low prevalence rates, making it challenging to gather comprehensive data. The strength of this report is the involvement of clin ical, geneticists, ENT specialists, and other spe cialized physicians who work together as part of an HHT Unit. This collaborative approach en sures the screening and diagnosis of HHT. Their expertise and knowledge in identifying HHT cases contribute to accurate and reliable diag noses. By ensuring that trained professionals are involved in the diagnostic process, the report can confidently provide reliable information. The reliable data obtained can then be entered into the database, enhancing the overall quality of the study and its findings. The consistency of our calculated prevalence with previous studies also supports the reliability of our results.
This is the first prevalence report in Latin America. The data towards the rest of the coun tries on the continent may not be extrapolable due to the heterogeneity of the population in these other Latin American countries.

