











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCION
El shunt portosistémico congénito (SP) es una malformación vascular que comunica circulación portal y sistémica, salteando el paso hepático. Es una entidad que puede cursar de forma asintomática o manifestarse clínicamente por complicaciones asociadas. En algunos casos puede requerir cierre quirúrgico o intravascular.
A continuación, se presenta un paciente de 30 días de vida, que cursa con hepatitis colestásica grave desde el nacimiento. Se estudian posibles diagnósticos diferenciales, siendo la etiología responsable un SP extrahepático. Es una entidad de baja incidencia, cuya presentación clínica grave en edad neonatal es poco frecuente.
CASO CLÍNICO
Recién nacido de término (39 semanas), con bajo peso para edad gestacional (2460 gramos, percentil 2), sin otros antecedentes perinatológicos de relevancia. Nació en San Luis, Argentina, donde se iniciaron los estudios por ictericia, coluria y acolia desde el nacimiento. Se constató en laboratorio hepatitis colestásica, anemia, plaquetopenia y coagulograma alterado (Tabla 1).
Como primer abordaje se realizaron:
• Serologías: negativas para adenovirus, citomegalovirus, virus Coxsackie B1 y B6, herpes 1 y 2, parvovirus, HIV, hepatitis B y C, toxoplasmosis, rubéola, Chagas y varicela.
• Ecografía con Doppler abdominal: hepatoesplenomegalia, sin otros hallazgos. En su lugar de origen recibió luminoterapia
y fenobarbital para tratamiento de la hiperbilirrubinemia, y ácido ursodesoxicólico por la colestasis. Por escasa respuesta y no arribar a un diagnóstico, se decidió la derivación a un centro de mayor complejidad.
Al momento de la derivación, el paciente tenía 31 días de vida. Se constató mal progreso ponderal, ictericia generalizada, coluria, acolia persistente y ligera tendencia a la irritabilidad. A la auscultación cardíaca, se evidenció soplo holosistólico a predominio en mesocardio. Al momento del ingreso, persistía la hepatitis colestásica con hiperamoniemia, aumento de ferritina, alfafetoproteína y ácidos biliares en sangre, anemia y plaquetopenia (Tabla 1).
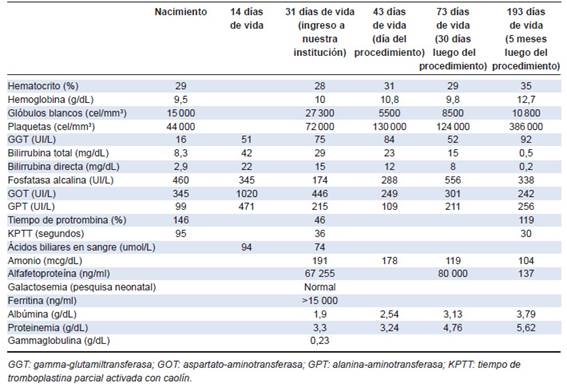
Tabla 1: Valores de laboratorio del paciente. Las dos primeras columnas corresponden a valores referidos del hospital de origen en San Luis; las restantes cuatro, a análisis realizados en nuestra institución
Se repitió la pesquisa neonatal con 6 determinaciones normales. Se evaluaron posibles causas de hepatitis colestásica: infecciosas, metabólicas, toxicológicas y anatómicas. Se obtuvieron los siguientes hallazgos de estudios complementarios:
• Ecocardiograma Doppler color: comunicación interauricular tipo ostium secundum, comunicación interventricular, ambos ventrículos dilatados, con contractilidad conservada.
• Ecografía abdominal con Doppler color: SP extrahepático (Figura 1 A): vaso venoso que comunica vena porta principal con vena cava inferior. Arteria hepática dilatada. Vena porta y ramas de pequeño tamaño. No se observaron alteraciones en la vía biliar.
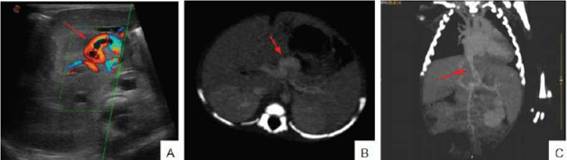
Figura 1: A. Ecografía Doppler que muestra comunicación anómala entre el sistema venoso portal y sistémico (flecha). 1 B y C. Tomografía computarizada, corte transversal (B) y coronal (C) que muestra la malformación (flecha)
• Angiotomografía de abdomen (Figuras 1B y C): conducto tortuoso (6,9 mm) que se originaba cefálico a la confluencia de las venas esplénica y mesentérica superior, y drenaba en vena cava inferior. Tronco de vena porta y sus ramas permeables. Arteria hepática aumentada de calibre (2,6 mm). Frente al diagnóstico de SP, con repercusión clínica y de laboratorio, se procedió al cierre de la malformación por angiografía digital. En la portografía se constató permeabilidad portal, se cateterizó el shunt y se embolizó con colocación de un plug de 4 mm, conservando flujo sanguíneo a través de este (Figura 2). Se decidió realizar un cierre parcial del defecto debido a la magnitud de la lesión, con intención de completar el cierre en un segundo tiempo. De esta forma, se minimizó el riesgo de aparición de complicaciones asociadas a la hipertensión portal derivada de la hipoplasia del sistema. Luego del procedimiento, el soporte nutricional se inició con nutrición parenteral total y progresión paulatina de la vía enteral, con el objetivo de preservar el flujo sanguíneo mesentérico. No se observaron complicaciones secundarias a hipertensión portal.

Figura 2: Angiografía digital que muestra el shunt con la flecha: previo (A) y luego (B) de la colocación del plug intravascular. Se observa pasaje de flujo sanguíneo a través de la comunicación vascular luego de la colocación del dispositivo (B)
En el seguimiento posterior, se observó mejoría en la clínica y en laboratorio, con disminución en los niveles de bilirrubina sérica (Figura 3). En la Tabla 1 se muestran los resultados de los análisis de sangre a los 30 días y 5 meses luego del procedimiento. Se destaca la persistencia de hepatitis, con elevación de transaminasas, gamma-glutamiltransferasa (GGT), fosfatasa alcalina y amonio, y normalización de la bilirrubina sérica, hematocrito y recuento de plaquetas.
A los 85 días de vida (42 días luego de la intervención), se constató por ecografía Doppler hepática plug normoposicionado en SP, sin flujo sanguíneo en su interior, venas portas permeables. A los 3 meses y medio, el paciente se encontró en condiciones de regresar a su lugar de origen.
En el seguimiento posterior, presentó mejoría en el progreso pondoestatural. Continúa en seguimiento por Neurología y Otorrinolaringología por presentar otoemisiones acústicas y potenciales evocados auditivos de tronco automatizados alterados.
DISCUSIÓN
En el desarrollo hepático fetal, existen numerosas conexiones vasculares intra- y extrahepáticas entre el sistema portal y sistémico, que posteriormente involucionan.
El SP congénito es una malformación vascular infrecuente que consiste en la persistencia de comunicaciones anómalas entre el sistema venoso portal y el sistémico.1 Pueden consistir en uno o más vasos, intra- o extrahepáticos, que derivan parcial o totalmente el flujo sanguíneo portal a la circulación sistémica (salteando el paso hepático) y se asocian a diversas complicaciones.2
Se estima una incidencia de 1:30 000/50 000 recién nacidos.1'3'4 El diagnóstico puede ser prenatal o posnatal en el contexto de un paciente en estudio por elevación de galactosemia en el cribado neonatal, por hepatitis colestásica, retraso en el crecimiento o por otras complicaciones asociadas. En ocasiones, puede ser un hallazgo incidental. Habitualmente, suele detectarse en edad pediátrica o adulta, mientras que su diagnóstico secundario a complicaciones presentes en etapa neonatal (como aquellas presentes en el caso clínico descrito) es más infrecuente.
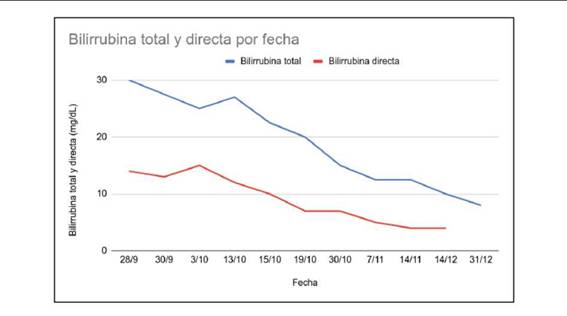
Figura 3: Gráfico que muestra valores séricos de bilirrubina total (en azul) y directa (en rojo). Se observa un descenso significativo desde el cierre de la malformación (10 de octubre)
Ante este diagnóstico, se debe realizar una búsqueda completa de otras malformaciones: alteraciones vasculares, renales, viscerales, esqueléticas y cardíacas.2-5 Las más frecuentemente descritas son comunicación interauricular o interventricular, ductus persistente, foramen oval permeable, coartación aórtica, tetralogía de Fallot y valvulopatías.6 Estas asociaciones sugieren una lesión prenatal en el desarrollo vascular cardíaco y abdominal, o presencia de cambios adaptativos en el corazón en respuesta al efecto hiperdinámico del SP. En el caso clínico, se describe como hallazgo comunicación interauricular e interventricular.
También se ha asociado a síndromes genéticos (por ejemplo: trisomía 21).3
Dentro de las complicaciones relacionadas al SP, se mencionan colestasis neonatal, insuficiencia hepática, poliesplenia con citopenias, tumores hepáticos, síndrome hepatopulmonar y encefalopatía hepática.
El diagnóstico se realiza a través de estudios imagenológicos, siendo la ecografía Doppler hepática de primera línea. Dentro de los hallazgos ecográficos posibles, Ponziani y cols.,6 describen como signos compatibles con la presencia de SP: 1) lesiones sólidas focales hepáticas; 2) vasos anómalos tortuosos que comunican el sistema portal y sistémico; 3) hipoplasia de vasculatura portal; 4) dilatación compensatoria de arteria hepática.
Para obtener mayor información sobre la anatomía de la lesión y el grado del desarrollo portal, se recomienda realizar una tomografía computada con contraste intravenoso.2 Se puede solicitar en su lugar una resonancia nuclear magnética, pero suele brindar menos información. La angiografía digital es útil para valorar la presencia de comunicaciones no detectables por otros métodos. A través de ella, se puede realizar la prueba de oclusión del shunt con balón para realizar la medición de presión portal (que refleja el grado de hipoplasia) y evaluar la tolerancia ante un eventual cierre total de la malformación.1'2 El valor de la presión en el sistema portal no debería superar los 18-25 mmHg.1
La necesidad de tratamiento se basa en dos características: la anatomía (dimensión y localización) y la presión medida en la prueba de oclusión. En aquellas malformaciones de menor calibre, intrahepáticas y no asociadas a complicaciones, se puede adoptar una conducta expectante, ya que pueden involucionar espontáneamente dentro de los 2 primeros años de vida.2 Los shunts de mayor tamaño, extrahepáticos, asociados a complicaciones o los que no han tenido un cierre espontáneo pueden requerir una intervención percutánea o quirúrgica para el cierre. En estas situaciones, se sugiere realizar la prueba de oclusión y, en caso de obtener presiones elevadas, realizar el cierre en dos tiempos para disminuir el riesgo de desarrollar hipertensión portal aguda luego del procedimiento.
En cuanto a la evolución a mediano y largo plazo, la plasticidad del sistema portal intrahepático permite la revascularización del hígado luego del cierre del shunt, incluso cuando no se detectan estructuras portales en estudios de imágenes previos. Esto deja poco o ningún lugar para el trasplante de hígado en el manejo de estos niños.
CONCLUSIÓN
El SP es una malformación poco frecuente, puede diagnosticarse como hallazgo en pacientes asintomáticos o asociado a complicaciones. El paciente del caso clínico, en la etapa neonatal, presentó múltiples manifestaciones asociadas al diagnóstico: hepatitis colestásica, ictericia, citopenias, coagulopatía y falla en el progreso pondoestatural. La presentación temprana es muy inusual, por lo que la sospecha diagnóstica es esencial para dirigir la búsqueda ecográfica (ya que su hallazgo puede ser difícil de evidenciar por un operador poco experimentado) y arribar al diagnóstico.
Recibido: 13-1-2022
Aceptado: 6-6-2022

