











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCION
Las microangiopatías trombóticas (MAT) se caracterizan por la presencia de anemia hemolítica microangiopática no inmune y trombocitopenia.1 Dentro de este grupo, se encuentra la púrpura trombocitopénica trombótica (PTT), una entidad rara en la edad pediátrica, potencialmente mortal y con una incidencia baja.2 Es causada por una grave deficiencia en la actividad de la proteasa ADAMTS13, definida por un nivel menor al 10 %; sin embargo, la deficiencia por sí sola no es suficiente para desencadenarla. En niños se presenta clínicamente con compromiso multiorgánico, alteraciones neurológicas, cardíacas, isquemia intestinal y, a veces, deterioro de la función renal y fiebre.1
Es importante tenerla en cuenta como diagnóstico diferencial de un cuadro de MAT por ser una patología con mortalidad alta sin tratamiento adecuado.1
CASO CLÍNICO
Paciente de sexo femenino de 15 años, previamente sana, que presentó cuadro de hemiparesia y parestesia braquiocrural derecha transitoria. Agregó posteriormente vómitos, dolor abdominal y astenia de 5 días de evolución. Al ingreso, se encontraba en buen estado general, eutrófica, ictérica, afebril, eucárdica, eupneica, con petequias generalizadas y hematoma en tobillo derecho. Presentaba abdomen blando, depresible e indoloro con el polo de bazo palpable, con examen neurológico sin datos positivos.
Al ingreso, se realizó un laboratorio que informó anemia normocítica normocrómica regenerativa con requerimiento transfusional (hemoglobina 5,4 mg/dl, volumen corpuscular medio 88, hemoglobina corpuscular media 29 y reticulocitos 28 %) y plaquetopenia grave (12 800/pL), con recuento y fórmula leucocitaria normales, asociado a alteración del hepatograma con hiperbilirrubinemia a predominio indirecto, elevación de aspartato-aminotransferasa (GOT) y lactato-deshidrogenasa (LDH), función renal normal. Se solicitó ecografía abdominal, tomografía computada, resonancia magnética nuclear y angiorresonancia de sistema nervioso central, que resultaron dentro de límites normales. Además, por un lado, se descartó patología autoinmune con laboratorio inmunológico normal y, por otro, infecciones agudas asociadas (HIV no reactivo, IgG citomegalovirus positivo,
IgG virus de la hepatitis C, IgM/IgG anti-core virus de la hepatitis B negativo, IgG herpes simple 1 y 2 positivo, VDRL y Chagas negativo). Comenzó seguimiento por hematología por sospecha de anemia hemolítica. Se ampliaron exámenes complementarios con prueba de Coombs, que resultó negativa, y frotis de sangre periférica que evidenció esquistocitos y signos de microangiopatía. Con estos criterios se diagnosticó MAT y, por alta sospecha de PTT, inició tratamiento inmediato con plasmaféresis y pulsos de metilprednisolona. Previamente, se solicitó determinación de ADAMTS13 que presentó actividad menor al 10 % con inhibidor positivo. Se diagnosticó púrpura trombótica trombocitopénica adquirida de causa autoinmune.
La paciente recibió 3 pulsos de metilprednisolona y ciclos de plasmaféresis, y normalizó valores de laboratorio. Por recaída de la enfermedad con plaquetopenia y aumento de parámetros de hemólisis a pesar del tratamiento instaurado en primera instancia, se decidió continuar con esquema de segunda línea con rituximab, del cual recibió 4 dosis.
Por mejoría clínica y hematológica, comenzó descenso de corticoides y continúa con controles ambulatorios de actividad de ADAMTS13.
DISCUSION
La PTT es una enfermedad rara, con alta mortalidad en ausencia de tratamiento oportuno. En niños presenta una incidencia de 0,1 casos por millón por año y fue descrita por primera vez en 1924.1'2 Predomina en el sexo femenino.
Se caracteriza por presentar anemia hemolítica no inmune, microangiopática y trombocitopenia por consumo. La fisiopatología está basada en el déficit grave de la actividad de la proteasa ADAMTS13. Este compromiso puede ser hereditario o adquirido por la formación de autoanticuerpos (el 10 % vs. el 90 % de los casos respectivamente).3 ADAMTS13 es una enzima necesaria para la proteólisis del factor de Von Willebrand (VWF) luego de su secreción por las células endoteliales; en ausencia del mecanismo regulatorio del tamaño multimérico del VWF, se genera trombosis plaquetaria microvascular, produciendo fragmentación de glóbulos rojos y consumo de plaquetas por la formación de trombos.1
El déficit de ADAMTS13 asociado a la ausencia de anticuerpos anti-ADAMTS13 sugiere PTT congénita, especialmente cuando se presenta en el período neonatal, en mujeres adultas durante el embarazo o en niños o en adultos con presentaciones típicas asociada a eventos gatillos y antecedentes familiares.4 El diagnóstico se confirma con la identificación de la mutación mediante el estudio genético molecular. La evolución clínica es heterogénea; puede cursar como forma crónica grave con requerimiento de tratamiento profiláctico mensual, o bien como forma leve a moderada con períodos de recaída y remisión.2
En la paciente presentada, observamos el laboratorio característico de anemia hemolítica, con signos de microangiopatía en el frotis y una actividad de ADAMTS13 menor al 10 % con anticuerpos IgG que neutralizan la actividad de proteólisis de ADAMTS13, por lo que fue categorizada como PTT adquirida. Esta variante, la más frecuente, se presenta clínicamente de manera aguda con tendencia a la recaída.1 Las alteraciones hematológicas asociadas a compromiso sistémico son la primera clave para establecer la sospecha, aunque no son específicas. Los principales órganos afectados suelen ser el sistema nervioso central, corazón y riñones.
El 60 % de los casos, como nuestra paciente, tiene manifestaciones neurológicas de gravedad variable, como parestesias, cefalea, alteraciones visuales, convulsiones, deterioro del sensorio e incluso coma. Puede haber compromiso cardíaco, como alteraciones electrocardiográficas inespecíficas, taquiarritmias, miocardiopatías y síndromes isquémicos; en este caso, la paciente presentaba trastornos inespecíficos de la repolarización en el electrocardiograma. El daño renal no es grave, debido a un probable efecto protector del ADAMTS13 local, por lo que, si existe disminución importante de la función renal, hay que descartar otras patologías. A nivel gastrointestinal, la isquemia intestinal puede manifestarse con dolor abdominal, vómitos y diarrea con sangre, y confundirse con un síndrome urémico hemolítico.
Los criterios diagnósticos de la PTT incluyen anemia hemolítica microangiopática y trombocitopenia sin otra causa que lo explique. Ante un cuadro clínico compatible, se deben solicitar hemograma con frotis de sangre periférica y prueba de Coombs, coagulograma, función renal y hepática, parámetros bioquímicos de hemólisis, enzimas cardíacas y beta-gonadotrofina coriónica humana (b-hCG) (si correspondiera según sexo y edad). Por otro lado, se deben estudiar serologías virales y realizar un perfil inmunológico, dada la prevalencia de enfermedades autoinmunes asociadas.
Una determinación de la actividad de ADAMTS13 menor al 10 % apoya el diagnóstico de PTT adquirida y el valor crítico de ADAMTS13 se asocia a mayor riesgo de recaída durante la evolución de la enfermedad, pero no es lo suficientemente sensible para confirmar el diagnóstico y tampoco específico para excluir pacientes con enfermedades subyacentes.3'5 La recuperación de la actividad de ADAMTS13 confirma el diagnóstico de PTT adquirida. Por otra parte, la presencia de un inhibidor funcional ADAMTS13 apoya, pero no confirma, el diagnóstico de PTT adquirida.5
La paciente del caso clínico cumplió con los criterios clínicos característicos, el laboratorio, el frotis de sangre periférica, la actividad de ADAMTS13 <10 % y la presencia de anticuerpos inhibidores de esta.
Como diagnósticos diferenciales de la PTT, se deben excluir otras microangiopatías trombóticas, como síndrome urémico hemolítico, anemia hemolítica inducida por drogas o infecciones, mediadas por complemento, desórdenes hereditarios del metabolismo de la vitamina B12, y otras enfermedades autoinmunes, como lupus eritematoso sistémico.
El tratamiento es una emergencia y el inicio temprano modifica el pronóstico y la evolución a largo plazo. La plasmaféresis y corticoides a altas dosis (prednisona 1 mg/kg/día o pulsos de metilprednisolona 30 mg/kg) se utilizan como primera línea.1 Antes del uso de la plasmaféresis, la mortalidad en pacientes con PTT adquirida era del 90 %,5 lo que obliga a iniciar tratamiento a todo paciente con anemia hemolítica microangiopática sin otra causa que lo justifique. Si no es posible realizar plasmaféresis, se debe comenzar con la infusión de 25-30 ml/kg de plasma fresco congelado hasta la derivación a institución donde se garantice el tratamiento adecuado.1
Se debe evaluar periódicamente la evolución una vez iniciado el tratamiento, considerando buena respuesta cuando aumenta el recuento plaquetario a 150 000/pL y permanece estable dos días consecutivos, normaliza el valor de LDH y hay mejoría o estabilidad de los síntomas neurológicos.1
Ante la falta de respuesta a medidas de primera línea, se debe continuar con terapias alternativas. El anticuerpo monoclonal anti-CD20, rituximab, se ha empleado con éxito en PTT refractaria y en recaída, con una tasa de respuesta del 87 % y del 100 % respectivamente.1 En el caso de nuestra paciente, por falta de respuesta favorable sostenida en el tiempo, luego del tratamiento inicial requirió el pasaje de rituximab semanal, con buena respuesta analítica (Figura 1).
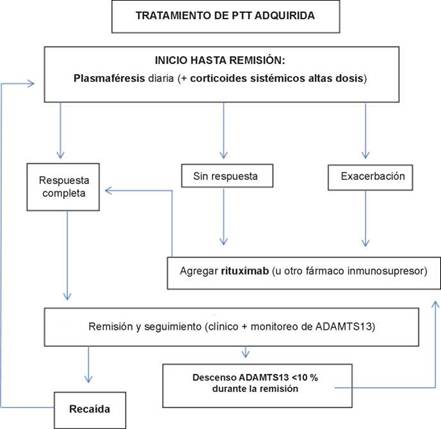
Figura 1: Tratamiento de púrpura trombótica trombocitopénica adquirida
En conclusión, la PTT adquirida es una enfermedad rara, pero con alta mortalidad y riesgo de secuelas sin tratamiento oportuno. En presencia de anemia hemolítica microangiopática y trombocitopenia sin otra causa que la justifique, debemos iniciar el tratamiento de forma inmediata.
Recibido: 29-6-2022
Aceptado: 3-11-2022

