










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO  uBio
uBio
Compartilhar

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versão impressa ISSN 0325-2957
Acta bioquím. clín. latinoam. vol.46 no.4 La Plata dez. 2012
CASO CLÍNICO
Paciente diabético de 54 años de edad con bajos niveles de colesterol sérico
A 54-year-old diabetic man with low serum cholesterol
Ugur Turk1, Gunes Basol2*, Burcu Barutcuoglu2, Fahri Sahin3,Sara Habif2, Patrizia Tarugi4,Oya Bayindir2
1 Department of Cardiology, Central Hospital, Izmir, Turkey; Departments of
2 Clinical Biochemistry and
3 Hematology, Ege University Faculty of Medicine, Izmir, Turkey;
4 Department of Biomedical Sciences, University of Modena e Reggio Emilia, Modena, Italy
* Dirección para correspondencia del autor: Ege University Faculty of Medicine, Department of Clinical Biochemistry, 35100 Izmir, Turkey.
Fax: _90-232-3392144;
e-mail gunes.basol@ege.edu.tr.
Traducción: Bioq. Leonardo Cacciagiú y Prof Dra Laura Schreier. Depart Bioquímica Clinica, FFyB-UBA
Este artículo ha sido traducido con el permiso de la AACC. La AACC no es responsable de la exactitud de la traducción. Las opiniones expresadas son las de los autores y no necesariamente de la AACC o de la Revista. Tomado de Clin Chem, 2012, 58 (5):826-830, con el permiso del editor. Derechos de autor original © Asociación Americana de Química Clínica, Inc, 2012. Al citar este artículo, por favor recurra a la fuente original de publicación en la revista Clinical Chemistry.
This article has been translated with the permission of AACC. AACC is not responsible for the accuracy of the translation. The views presented are those of the authors and not necessarily those of the AACC or the Journal. Reprinted from Clin Chem, 2012 58(5): 826-830, by permission of the publisher. Original copyright © 2012 American Association for Clinical Chemistry, Inc. When citing this article, please refer to the original publication source in the journal, Clinical Chemistry.
Este caso fue presentado como una presentación de póster en el IFCC-WorldLab y EuroMedLab, Berlín 2011. Recibido el 09 de febrero 2011, aceptado el 20 de julio 2011. Doi: 10.1373/clinchem.2011.163543
Descripción del caso
Paciente de sexo masculino, de 54 años de edad, asintomático, con historia de diabetes mellitus tipo 2 (T2DM)5 que desde 5 años atrás, mostró una concentración sérica de colesterol muy baja. No tenía antecedentes de enfermedad grave en la infancia, síndrome de mala absorción, o cualquier disfunción cardiovascular o neurológica. Fue fumador durante 30 años y no consumía alcohol ni fármacos hipolipemiantes. Además, no era vegetariano. Su historia familiar incluye accidente cerebrovascular (padre fallecido a los 52 años) y enfermedad renal crónica (hermano de 57 años de edad). Su hijo mayor había muerto de un presunto infarto de miocardio a los 21 años. El paciente presentaba una presión arterial de 120/80 mmHg, una frecuencia cardiaca de 78 latidos/min, y un índice de masa corporal de 32 kg/m2. El examen físico fue normal. Se observó esteatosis hepática y hepatomegalia leve a través de ecografía abdominal. El ecocardiograma fue normal, y los resultados de la ergometría (protocolo de Bruce) fueron negativos.
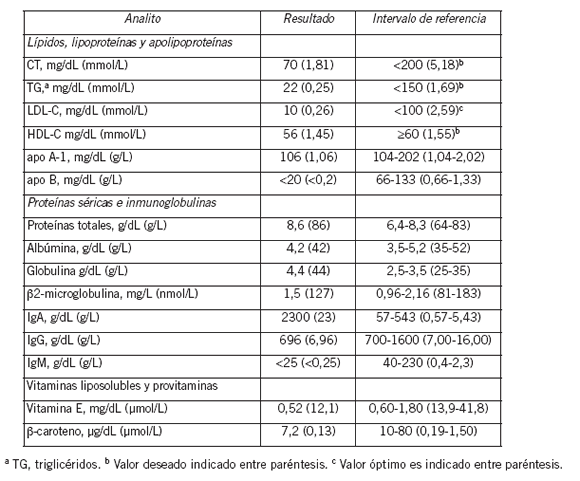
Se llevaron a cabo estudios de laboratorio. Los resultados séricos de enzimas hepáticas, pruebas de función tiroidea y valores de los parámetros hematológicos fueron todos normales, así como los de bilirrubina, creatinina, urea, ácido úrico y calcio. La glucosa en ayunas se encontró incrementada [155 mg/dL (8,6 mmol/L), intervalo de referencia, 60-110 mg/dL (3,33-6,11 mmol/L)], y el valor de la hemoglobina A1c fue de 7% (intervalo de referencia, 4%-6%). Los lípidos, lipoproteínas, apolipoproteínas, proteínas, inmunoglobulinas, vitaminas liposolubles y provitaminas se muestran en la Tabla I. Es de destacar que las concentraciones séricas de colesterol total (CT), triglicéridos, colesterol-LDL (LDL-C) y apolipoproteína B (apo B) se encontraban marcadamente disminuidas. Las concentraciones de proteína totales y globulinas fueron ambas altas. Los resultados de las pruebas serológicas para hepatitis viral A, B y C y para HIV fueron negativos.
Tabla 1. Resultados de laboratorio del paciente seleccionados con su correspondiente intervalo de referencia.
Preguntas a considerar
1. ¿Cuáles son las alteraciones lipídicas típicas que se observan en personas con diabetes tipo 2?
2. ¿Cuáles son las posibles causas de colesterol sérico, LDL y ApoB bajos?
3. ¿Qué otras pruebas se podrían hacer para aclarar la causa de la disminución de las concentraciones de lípidos en este caso?
4. Dado los resultados de las proteínas séricas totales y de las globulinas del paciente, ¿qué otras pruebas se deberían realizar?
Discusión
Reseña de hipobetalipoproteinemia
La hipobetalipoproteinemia (HBL) se define por concentraciones plasmáticas de colesterol total (CT), LDL-C y apo B más bajas que el percentilo quinto (1). HBL primaria incluye a un grupo de trastornos genéticos: abetalipoproteinemia (ABL), enfermedad de retención de quilomicrones (CMRD), y HBL familiar (FHBL). ABL y CMRD son trastornos muy raros causados por mutaciones recesivas en los genes MTTP6 (proteína microsomal de transferencia de triglicéridos) y SAR1B [SAR1 homólogo B (S. cerevisiae)] respectivamente. La ABL es una condición que generalmente se diagnostica temprano en la vida y se asocia con esteatorrea, intolerancia oral grasa, acantocitosis, retinitis pigmentaria y alteraciones neurológicas. El perfil lipídico de los pacientes con ABL se caracteriza por una concentración plasmática extremadamente baja de CT, VLDL y LDL, y una ausencia casi total de apo B. La CMRD se caracteriza por la ausencia de apo B-48 en plasma. Sus principales manifestaciones clínicas son esteatorrea, malnutrición y retraso del crecimiento. Debido a que la síntesis hepática de apo B se mantiene, las LDL están presentes en el plasma. FHBL es un trastorno codominante con una frecuencia, en la forma heterocigota, de 1 en 500 a 1 en 1000. Las formas heterocigotas de la FHBL suelen ser asintomáticas, pero también pueden presentar hígado graso no alcohólico y un leve aumento en las enzimas hepáticas. Los homocigotas para FHBL pueden experimentar severa malabsorción de grasa y muestran graves manifestaciones clínicas y bioquímicas similares a las de ABL. Curiosamente, las concentraciones plasmáticas de apo B son menores a lo esperado para la deficiencia de sólo 1 gen, como el presente caso. Aproximadamente 50% de los pacientes FHBL son portadores de mutaciones patológicas en el gen APOB [apolipoproteína B (incluyendo el antígeno Ag (x)]. La mayoría de las mutaciones del gen APOB causan la formación de formas truncadas de apo B, que reducen la capacidad para exportar los lípidos de los hepatocitos como constituyentes lipoproteicos. Las formas truncadas de apo B con un tamaño menor que apo B-30 no son detectables en plasma porque se eliminan rápidamente. En pacientes con moderada HBL parecen ser más frecuentes las formas truncadas detectables, porque muchas moléculas largas de apo B truncadas mantienen una capacidad de unión residual de lípidos para formar partículas de lipoproteínas. Las moléculas de apo B truncadas más cortas que la apo B-70.5 se eliminan del plasma mayormente por riñón, mientras moléculas de apoB truncadas con un tamaño mayor del 70% de la apoB-100 son eliminadas por el hígado (1)(2).
La HBL también puede ser causada por varios factores no genéticos, tal como una dieta vegetariana estricta, malnutrición, drogas, y condiciones relacionadas con la enfermedad. Estos factores son considerados como causas secundarias de la HBL. Dado que el hígado desempeña un papel clave en el metabolismo de la mayoría de lipoproteínas y apolipoproteínas plasmáticas, se pueden observar alteraciones en el patrón lipídico plasmático en condiciones caracterizadas por daño celular hepático, tales como infecciones por virus de la hepatitis B y C, cirrosis o carcinoma hepatocelular. Las enfermedades crónicas del parénquima hepático (incluyendo carcinoma hepatocelular) conducen a una disminución en el colesterol plasmático al afectar la síntesis y el metabolismo del colesterol. Por otra parte, el aumento del consumo de colesterol por las células tumorales influye en la reducción sérica del colesterol en el carcinoma hepatocelular (3). La infección por HIV en etapa avanzada se caracteriza por una reducción de CT, HDL-C, y LDL-C y concentraciones incrementadas de triglicéridos en el plasma (4). El aumento de la excreción del colesterol y un mayor recambio de LDL serían responsables de la hipocolesterolemia observada en el hipertiroidismo (5). En el caso de pacientes renales crónicos en hemodiálisis, la desnutrición y la inflamación serían las responsables de la hipocolesterolemia frecuente (6).
Análisis adicional de los bajos niveles de ldl-c y apo b
La diabetes tipo 2 suele acompañarse de hiperlipoproteinemia secundaria con aumento de VLDL-C y disminución de HDL-C. Es importante destacar que a pesar de la presencia de T2DM, el paciente en este caso presenta concentraciones de lípidos muy bajas en suero. Cuando se indagó más al paciente, recordó que previamente había tenido bajas concentraciones de colesterol (datos no disponibles). Para descartar posibles interferencias en la medición, se analizó la cinética de reacción de los parámetros lipídicos que se habían medido con el Sistema modular de Roche y kits comerciales. Las curvas de las determinaciones fueron normales, evay los datos del paciente cumplieron con todos los criterios bioquímicos para el diagnóstico de HBL.
En el diagnóstico diferencial, primero fueron excluidas las causas secundarias de HBL. El paciente no era vegetariano y no utilizaba fármacos hipolipemiantes. Además, no tenía signos, síntomas o hallazgos de laboratorio de cualquier enfermedad que pudiera asociarse con HBL secundaria. Por lo tanto, la enfermedad del paciente fue diagnosticada fenotípicamente como HBL primaria.
La ABL, CMRD, y FHBL homocigota se encuentran asociados con un fenotipo clínico grave, en particular en niños y jóvenes adultos (2). Nuestro paciente, sin embargo, no mostró evidencia de malabsorción, retinitis pigmentosa o enfermedad neurológica, y tampoco había evidencias de acantocitos. El fenotipo clínico leve sugiere fuertemente el diagnóstico clínico de FHBL heterocigoto. Teniendo en cuenta que la historia familiar del paciente incluyó muerte súbita de un hijo joven y que los portadores de FHBL heterocigoto con formas truncadas cortas, ante la presencia de factores que pueden causar lesión hepática, estarían en riesgo de desarrollar enfermedad hepática más severa. Confirmamos el diagnóstico mediante la identificación de la mutación en el gen APOB. El análisis de secuencia del gen APOB mostró la presencia de una sustitución de un sólo nucleótido en el exón 26 (c.7692C>T) en el estado heterocigoto. Esta sustitución convierte el codón de arginina en la posición 2495 en un codón de terminación (p.R2495X), conduciendo a la formación de una apo B truncada que contiene 2494 residuos de aminoácidos (en lugar de los 4536 residuos presentes en la proteína completa de apo B). Esta apo B truncada, designada apo B-55 de acuerdo con la nomenclatura aceptada, ha sido previamente descrita en FHBL (7)( 8).
FHBL, Diabetes tipo 2 y enfermedad cardiovascular
La enfermedad cardiovascular es una severa complicación asociada a la T2DM. Resultados prospectivos del estudio Bruneck mostraron que la T2DM es un fuerte predictor independiente de aterosclerosis carotídea avanzada (9). El uso del espesor de la íntima-media carotídea como marcador sustituto de enfermedad cardiovascular se ha aplicado en varios estudios que investigaron pacientes con T2DM. Aunque nuestro paciente tenía varios factores de riesgo cardiovascular, no manifiestó ningún tipo de complicación macrovascular. Además, el espesor de la íntima-media carotídea (0,53-0,58 mm) fue normal y sin placa aterosclerótica. Estos hallazgos sugieren un efecto protector de los bajos niveles de LDL-C en FHBL y son compatibles con aquellos publicados por Pulai et al., que no encontraron complicaciones macrovasculares en dos portadores de apo B-55 con T2DM (8).
Evaluación del aumento de proteínas totales del suero
El aumento en el paciente de las concentraciones séricas de proteínas totales y globulinas requerirá mayor evaluación. La cuantificación de inmunoglobulinas mostró un gran aumento de la concentración de IgA, IgM marcadamente disminuida, e IgG levemente por debajo de límite inferior del valor de referencia (Tabla I). La electroforesis de proteínas séricas reveló un pico monoclonal de 1,57 g/dL (15,7 g/L) en la región ß. Esta banda monoclonal se caracterizó a través de electroforesis con inmunofijación sérica como banda IgAκ. En la orina la electroforesis de proteínas con inmunofijación no reveló la presencia de una banda monoclonal. Las células plasmáticas aumentaron ligeramente (entre 5%-6%) en la médula ósea (intervalo referencia, 0,2%-2,2%). El estudio radiológico del esqueleto no reveló lesiones osteolíticas. Debido a que el paciente no presentó manifestaciones clínicas relacionadas con gammopatía monoclonal, tales como hipercalcemia, anemia, insuficiencia renal o lesiones óseas, se le diagnosticó una gammopatía monoclonal de significado incierto (MGUS). La MGUS es un trastorno pre-maligno asintomático, que se identifica mediante análisis de sangre de rutina, por lo general en personas mayores a 50 años de edad. La MGUS se define por una concentración de proteína monoclonal en suero ≤3,0 g/dL (≤30 g/L) y por ≤10% de células plasmáticas en la médula ósea sin evidencia de mieloma múltiple o de otro trastorno maligno relacionado (10).
Por lo tanto, hemos identificado la FHBL y la MGUS, independientemente una de la otra, en el paciente asintomático con diabetes, que fue remitido para una evaluación adicional por su muy baja concentración sérica de colesterol. El paciente fue derivado a los servicios de endocrinología y gastroenterología para el seguimiento de la T2DM e hígado graso, respectivamente. Además, el servicio de hematología también comenzó a monitorearlo, ya que los pacientes con MGUS tienen mayor riesgo de progresión a mieloma múltiple.

Contribuciones de los autores: Todos los autores confirmaron que han contribuido al contenido intelectual de este documento y han cumplido con los siguientes 3 requisitos: (a) contribuciones significativas a la concepción y el diseño, adquisición de datos, o análisis e interpretación de datos, (b) la redacción o la revisión del artículo de contenido intelectual, y (c) la aprobación final del artículo publicado.
Declaraciones de los autores o los posibles conflictos de interés: Ningún autor declaró algún potencial conflicto de interés.
5 Abreviaturas no usuales: T2DM, diabetes mellitus tipo 2; CT, colesterol total; LDL-C, colesterol LDL; apo B, apolipoproteina B; HBL, hipobetalipoproteinemia; ABL, abetalipoproteinemia; CMRD, enfermedad de retención de quilomicrones; FHBL, hipobetalipoproteinemia familiar; MGUS, gammopatía monoclonal de significado incierto
6 Genes humanos: MTTP, proteína microsomal de transferencia de triglicéridos; SAR1B, SAR1 homologo B (S. cerevisiae); APOB, apolipoproteina B (incluyendo antígeno x, Agx).
1. Schonfeld G. Familial hypobetalipoproteinemia: a review. J Lipid Res 2003; 44: 878-83.
2. Tarugi P, Averna M, Di Leo E, Cefalu AB, Noto D, Magnolo L, et al. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis 2007; 195: e19-27.
3. Jiang J, Nilsson-Ehle P, Xu N. Influence of liver cancer on lipid and lipoprotein metabolism. Lipids Health Dis 2006; 5:4.
4. Grunfeld C, Pang M, Doerrler W, Shigenaga JK, Jensen P, Feingold KR. Lipids, lipoproteins, triglyceride clearance, and cytokines in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. J Clin Endocrinol Metab 1992; 74: 1045-52.
5. Duntas LH. Thyroid disease and lipids. Thyroid 2002; 12: 287-93.
6. Kaysen GA. Biochemistry and biomarkers of inflamed patients: why look, what to assess. Clin J Am Soc Nephrol 2009; 4(Suppl 1): S56-63.
7. Welty FK, Ordovas J, Schaefer EJ, Wilson P, Young SG. Identification and molecular analysis of two apoB gene mutations causing low plasma cholesterol levels. Circulation 1995; 92: 2036-40.
8. Pulai JI, Latour MA, Kwok PY, Schonfeld G. Diabetes mellitus in a new kindred with familial hipobetalipoproteinemia and an apolipoprotein B truncation (apoB-55). Atherosclerosis 1998; 136: 289-95.
9. Bonora E, Kiechl S, Oberhollenzer F, Egger G, Bonadonna RC, Muggeo M, Willeit J. Impaired glucosa tolerance, type II diabetes mellitus and carotid atherosclerosis: prospective results from the Bruneck Study. Diabetologia 2000; 43: 156-64.
10. Kyle RA, Rajkumar SV. Monoclonal gammopathies of undetermined significance. Best Pract Res Clin Haematol 2005; 18: 689 -707.
Comentarios
Mohit Jain1 and Jorge Plutzky1 *
En este caso de hipobetalipoproteinemia (HBL), se ha proporcionado el diagnóstico diferencial para HBL genética y secundaria. Se pueden observar varios problemas adicionales.
Este paciente también presentaba una paraproteinemia a IgA, que puede influir en las concentraciones de colesterol. La paraproteinemia monoclonal obstaculizaría el aclaramiento de lipoproteínas, que aunque aumente el colesterol circulante, se producen artefactos que reducen la medición de colesterol (1). En este paciente las concentraciones de colesterol fueron inferiores a las típicamente observadas con HBL primarias heterocigotas, adjudicado a la cuestión de la interferencia por la paraproteína o a otra mutación heterocigota no identificada que influiría en la concentración de apolipoproteína B (apo B). Tal vez, en la muerte del hijo de 21 años de edad estaría en juego otra variante familiar agregada En la hipocolesterolemia familiar ha recibido reciente atención la identificación de mutaciones en los genes ANGPTL32 (angiopoyetina tipo-3) y PCSK9 (proproteína convertasa subtilisina/kexina tipo 9) como causas novedosas de concentraciones bajas de colesterol (2). Tal como las variantes de apo B asociadas a HBL, las mutaciones en ANGPTL3 y PCSK9 parecen ser bien toleradas y potencialmente ateroprotectoras, características que generan interés como objetivos terapéuticos. En esta línea, se encuentra en fase final de desarrollo terapéutico, la inhibición de la transcripción del gen APOB utilizando oligonucleótidos antisentido.
Los intentos en marcha para reducir las concentraciones de apo B, a pesar del éxito de las estatinas y otros medicamentos reductores del colesterol (por ejemplo, ezetimibe, secuestrantes de ácidos biliares, niacina), reflejan muchas cuestiones clínicas: intolerancia a las estatinas, altas concentraciones de colesterol, reducción de las metas de LDL, y la identificación cada vez mayor de hipercolesterolemia familiar y sus desafíos de tratamiento.
1 Vascular Disease Prevention Program, Cardiovascular Division, Brigham and Women's Hospital, Boston, MA.
* Dirección de correspondencia del autor: Vascular Disease Prevention Program, Cardiovascular Division, Brigham and Women's Hospital/Harvard Medical School, 77 Avenue Louis Pasteur, NRB 742, Boston, MA 02115. Fax 617-525-4366; e-mail jplutzky@rics.bwh.harvard.edu.
Recibido Febrero 22, 2012;
Aceptado Febrero 28, 2012.
DOI: 10.1373/clinchem.2012.182139
2 Genes humanos: ANGPTL3, simil-angiopoyetina 3; PCSK9, proproteína convertasa subtisilina/kexina tipo 9.
Contribuciones de los autores: Todos los autores confirmaron que han contribuido al contenido intelectual de este documento y han cumplido con los siguientes 3 requisitos: (a) contribuciones significativas a la concepción y el diseño, adquisición de datos, o análisis e interpretación de datos, (b) la redacción o la revisión del artículo de contenido intelectual, y (c) la aprobación final del artículo publicado.
Declaraciones de los autores o los posibles conflictos de interés: Ningún autor declaró algún potencial conflicto de interés.
1. Tsai LY, Tsai SM, Lee SC, Liu SF. Falsely low LDL-cholesterol concentrations and artifactual undetectable HDL-cholesterol measured by direct methods in a patient with monoclonal paraprotein. Clin Chim Acta 2005; 358: 192-5.
2. Calandra S, Tarugi P, Speedy HE, Dean AF, Bertolini S, Shoulders CC. Mechanisms and genetic determinants regulating sterol absorption, circulating LDL levels, and sterol elimination: implications for classification and disease risk. J Lipid Res 2011; 52: 1885-926.
Comentario
1 Department of Laboratory Medicine, Nacional Institutes of Health, Bethseda, MD.
* Dirección de correspondencia del autor: Department of Laboratory Medicine, Nacional Institutes of Health, Bldg 10, Rm. 2C-433, Bethesda, MD 20892. Fax 301-402-1885; e-mail aremaley@cc.nih.gov.
Recibido Febrero 16, 2012;
Aceptado Febrero 21, 2012.
DOI: 10.1373/clinchem.2012.182147.
Este caso clínico estudiado ilustra muy bien los problemas típicos que se encuentran al tratar de entender la causa subyacente de un paciente con un fenotipo lipídico anormal. Tal como se hizo en este caso, es muy importante tratar de obtener la mayor información posible acerca de los valores de lípidos de otros miembros de la familia. Los pacientes que son homocigotas para hipobetalipoproteinemia familiar (FHBL) por lo general tienen algún padre con bajos valores de apolipoproteína B (apo B). En cambio, los padres de los pacientes con abetalipoproteinemia generalmente tienen concentraciones normales de apo B, debido a que es un trastorno autosómico recesivo. En este caso, la ausencia de historia de malabsorción y la detección de una sola mutación de apo B son consistentes con la FHBL heterocigota. Estos pacientes frecuentemente tienen concentraciones de apo B menos de la mitad de lo normal, lo cual probablemente sea una consecuencia de la disminución del tamaño del pool de apo B y del buen catabolismo de LDL, pero a diferencia de este caso, la apo B es aún detectable. En este reporte no está claro cómo fue secuenciado el gen de APOB. Si sólo se examinaron los exones, el paciente tal vez tiene una mutación en el promotor del intrón o del otro alelo que conduce a la expresión disminuida. Debido a que la mutación descubierta de apo B es corriente abajo del sitio de edición de apo B, la síntesis de apo B-48 para la producción de quilomicrones puede haber estado conservada, evitando así la malabsorción. El paciente también podría tener una mutación en otro gen que afecta el metabolismo de apo B, tal como el PCSK92 (proproteína convertasa subtilisina/kexina tipo 9), lo que puede reducir las concentraciones de apo B modulando la actividad del receptor de LDL (1). Otra posibilidad es que la paraproteína producida por la gammopatía monoclonal de significado incierto podría estar involucrada. Los autoanticuerpos contra los componentes proteicos o lipídicos de las lipoproteínas podrían disminuir el colesterol LDL y HDL; tales efectos se pueden examinar con estudios mixtos (2). Sin embargo, este es un caso interesante de una enfermedad rara que es importante reconocer para que se pueda iniciar una terapia con altas dosis de vitaminas A y E. El tratamiento puede prevenir problemas neurológicos graves y de visión que pueden desarrollar los pacientes con FHBL homocigotas y con abetalipoproteinemia, por la disminución del transporte de vitaminas liposolubles en las lipoproteínas que contienen apo B.
Contribuciones de los autores: Todos los autores confirmaron que han contribuido al contenido intelectual de este documento y han cumplido con los siguientes 3 requisitos: (a) contribuciones significativas a la concepción y el diseño, adquisición de datos, o análisis e interpretación de datos, (b) la redacción o la revisión del artículo de contenido intelectual, y (c) la aprobación final del artículo publicado.
Declaraciones de los autores o los posibles conflictos de interés: Ningún autor declaró algún potencial conflicto de interés.
2 Genes humanos: PCSK9, proproteína convertasa subtilisina/kexina tipo 9.
1. Horne MK 3rd, Merryman P, Cullinane A, Remaley AT. In vitro characterization of a monoclonal IgG(kappa) from a patient with planar xanthomatosis. Eur J Haematol 2008; 80: 495-502.
2. Tibolla G, Norata GD, Artali R, Meneghetti F, Catapano AL. Proprotein convertase subtilisin/kexin type 9 (PCSK9): from structure-function relation to therapeutic inhibition. Nutr Metab Cardiovasc Dis 2011; 21: 835-43.

