











 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkPresentamos el caso de un paciente de 54 años, con el único antecedente de trasplante hepático en 2019 por cirrosis alcohólica. Como parte del estudio pretrasplante, por el dato de hipertensión pulmonar (HTP) moderada en la ecocardiografía, se realiza un cateterismo derecho, que confirma que se trata de una HTP por hiperflujo, con resistencias pulmonares bajas.
A los tres meses postrasplante, el paciente acude a nuestro servicio de urgencias por disnea progresiva y signos de congestión pulmonar y periférica, aunque estable con oxigenoterapia con cánula nasal. Se realiza un electrocardiograma, que revela un bloqueo de rama derecha de nueva aparición. En la analítica, elevación de dímero D (2980 ug/mL) y del NT-proBNP, (10 500 ng/L). Para descartar la sospecha inicial de tromboembolismo pulmonar, se solicita una angiotomografía computada de arterias pulmonares (Figura 1), que no objetiva trombos, pero sí destaca una dilatación significativa de la arteria pulmonar, sugestiva de HTP significativa. Dada la escasa mejoría con diuréticos, es valorado por el servicio de Cardiología de nuestro centro. En la ecocardiografía transtorácica (ETT) (Figura 2), se distingue una dilatación y disfunción marcada del ventrículo derecho (VD), con una presión sistólica de la arteria pulmonar (PSAP) estimada de 120 mmHg, sugestivo de HTP grave.
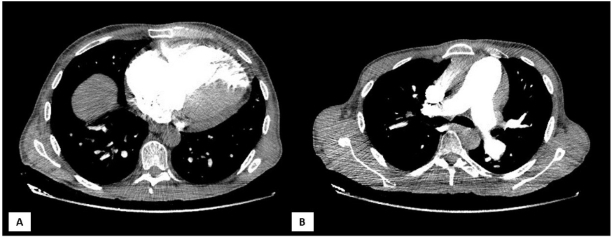
Fig. 1 Angiotomografía computada de arterias pulmonares. Cortes axiales a nivel de cavidades cardíacas ventriculares (A) y a nivel de salida de grandes vasos y bifurcación de la arteria pulmonar (B). No se aprecian defectos de repleción a nivel intravascular en la arteria pulmonar ni imágenes sugestivas de trombo (B). Llama la atención la dilatación de VD, con una relación ventrículo derecho/ventrículo izquierdo >1 (A). VD: ventrículo derecho

Fig. 2 Ecocardiograma transtorácico al ingreso. Planos apical 4 cámaras (A), paraesternal eje largo modificado sobre VD (B), paraesternal eje corto a nivel de grandes vasos (C) y Doppler continuo sobre flujo de insuficiencia tricuspídea (D). En (A) se aprecia dilatación y disfunción grave del VD, con insuficiencia tricuspídea grave funcional (B). Dilatación de la arteria pulmonar (C). El gradiente VD-AD de la insuficiencia tricuspídea por Doppler es de 120 mmHg (D), sugestivo de HTP grave. AD: aurícula derecha; HTP: hipertensión pulmonar; VD: ventrículo derecho
Dado el rápido empeoramiento clínico, sobre todo a nivel respiratorio, con mayor taquipnea y datos de insuficiencia cardiaca, se decide el ingreso en la Unidad Coronaria, donde se inicia tratamiento endovenoso con dosis crecientes de dobutamina y furosemida (1 gramo en infusión continua). Se realizan asimismo paracentesis evacuadoras, para manejo sintomático de la ascitis, y se comprueba por eco Doppler el correcto funcionamiento del injerto hepático.
Ante un paciente con antecedentes de cirrosis, que se presenta con un cuadro de disnea e insuficiencia cardiaca aguda, el diagnóstico diferencial a plantearnos es amplio.
El síndrome porto-pulmonar (PoPH) se engloba dentro de la HTP grupo 1, 1 y constituye el 10% del total de casos de esta entidad. Se define como la presencia de hipertensión pulmonar arterial (HAP) asociada a hipertensión portal. 2 Ocurre en el 1-2% de estos pacientes; 1 de hecho en el Registro REVEAL, 3 estudio observacional multicéntrico de 3000 pacientes con HAP, la prevalencia del PoPH fue del 5%, más frecuente en mujeres y en cirrosis de causa autoinmune. La gravedad se define en función del valor de la presión pulmonar media (PAPm), de modo que se habla de PoPH leve en pacientes con PAPm <35 mmHg, y PoPH grave en caso de PAPm >45 mmHg. La patogénesis de esta entidad no está bien aclarada, si bien se piensa que se debe a un desbalance entre mediadores vasoconstrictores y vasodilatadores. La mayoría de los pacientes se encuentran asintomáticos; entre los que tienen síntomas el más frecuente es la disnea de esfuerzo, como el caso de nuestro paciente.
Las guías europeas actuales más recientes 1 recomiendan iniciar el estudio diagnóstico con el ETT, de modo que en aquellos con datos indirectos de HTP (velocidad pico de la insuficiencia tricuspídea > 2,8 m/s, dilatación del VD, dilatación de la arteria pulmonar o de la vena cava inferior) o con otros factores de riesgo, se indicaría la realización de un cateterismo derecho, tal como se hizo en nuestro paciente. El estudio reveló una HTP grave precapilar: PAPm 57 mmHg, presión capilar pulmonar (PCP) 11mmHg, gradiente transpulmonar elevado, de 46 mmHg, y resistencia vascular pulmonar (RVP) de 14,5 unidades Wood.
Los criterios diagnósticos hemodinámicos actuales 1 para concluir HAP son: PAPm >20 mmHg en re poso, PCP ≤15mmHg y RVP >2 UW. 1
El último criterio para el diagnóstico de certeza de PoPH sería confirmar la presencia de hipertensión portal, generalmente por clínica, siendo posible en caso de duda la realización de un cateterismo venoso para medir el gradiente venoso hepático. 4
En el caso de nuestro paciente, este último criterio no se cumple, por lo que se lo etiqueta como “probable síndrome portopulmonar”, si bien se han descrito casos aislados en la bibliografía de diagnóstico de novo de PoPH en los primeros 6 meses postrasplante.
Se trata de una entidad de mal pronóstico: sin tratamiento, se asocia a una supervivencia del 14% a los 5 años, 2 si bien se ha descrito una mejoría de la supervivencia a 5 años hasta el 51% con tratamiento médico, y hasta el 81% con trasplante hepático. 5
Pese a que se han publicado resultados favorables con el tratamiento médico de la HAP, la mayoría de estudios no van dirigidos a pacientes con PoPH (salvo el estudio PORTICO que demostró resultados favorables a nivel hemodinámico con macitentán; o el estudio PATENT-1, 6 que incluyó una pequeña proporción de pacientes con PoPH, con buenos resultados funcionales con riociguat); en todo caso, se ha visto que esta terapia tiene efectos favorables a nivel hemodinámico y funcional, pero sin efecto en la supervivencia.
Las guías actuales 1 recomiendan, en casos de riesgo elevado, como en nuestro paciente, comenzar directamente con triple terapia con un antagonista de receptor de la endotelina, un inhibidor de la fosfodiesterasa-5 y un agonista de la vía de prostaciclinas (recomendación clase IIa). Se decide en nuestro paciente comenzar con epoprostenol, sildenafil y macitentán, y se consigue una lenta pero progresiva mejoría, con retiro de los inotrópicos.
El trasplante hepático clásicamente se ha considerado contraindicado en estos pacientes, dada la elevada morbimortalidad perioperatoria, y solamente estaría indicado en pacientes con indicación de trasplante por su hepatopatía per se; en el caso de nuestro paciente, estando el injerto normofuncionante, no era algo a plantearse. Las guías recomiendan iniciar tratamiento de la HAP si la PAPm es >35 mmHg en pacientes que sean candidatos a trasplante, 3 que es contraindicado en pacientes con PoPH grave que no mejore con tratamiento médico, ya que la mortalidad perioperatoria en pacientes con PAPm>45 mmHg se acerca al 100%.
Tras un mes de ingreso, el paciente es dado de alta, con mantención de la triple terapia, en control por hospitalización a domicilio. En el ETT de control a los 6 meses, se aprecia cómo la función del VD ha normalizado, y se pueden retirar progresivamente los fármacos para su HAP.
El diagnóstico diferencial ha de plantearse con la HTP por hiperflujo (con RVP normal, sin que sea necesario iniciar tratamiento específico) y con el síndrome hepatopulmonar (que clásicamente cursa sin HTP, se caracteriza por shunts arteriovenosos en la circulación pulmonar, que ocasionan hipoxemia, ortodesoxia y pla tipnea; siendo el tratamiento de elección el trasplante hepático en casos graves).
Otras entidades menos comunes, pero que no hemos de olvidar en pacientes con antecedente de hepatopatía son la cardiopatía cirrótica, otras causas de insuficien cia cardiaca clásicas, y causas extracardíacas de disnea, comunes en este perfil de paciente, como anemia, ascitis, o hidrotórax.
En conclusión, la disnea en el paciente con antecedentes de hepatopatía clásicamente ha sido un reto para el cardiólogo, no solo por el amplio abanico de diagnósticos a valorar, sino también por su complejo perfil hemodinámico. El síndrome portopulmonar constituye una entidad poco frecuente. Pese a que en su definición clásica se contempla en pacientes con hipertensión portal, es una entidad que comienza a describirse en pacientes ya trasplantados, especialmente de forma precoz en los primeros 6 meses postrasplante, tal como reflejamos en este caso, por lo que hemos de tenerla siempre en cuenta, dado su mal pronóstico y la ausencia de un tratamiento específico per se.

