











 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkWe report the case of a 54-year-old male patient with a history of liver transplantation in 2019 due to alcoholic cirrhosis. A right heart catheterization was performed as part of the pretransplant evaluation, due to moderate pulmonary hypertension (PH) on echocardiography, confirming that PH was associated with high pulmonary blood flow and low pulmonary resistances.
Three months after transplantation, the patient came to our emergency room for progressive dyspnea and signs of pulmonary and peripheral congestion, but stable with nasal cannula oxygen therapy. ECG showed new-onset right bundle branch block. Laboratory tests revealed elevated D-dimer (2980 ug/mL) and NT-proBNP (10 500 ng/L) levels. A computed angiotomography of the pulmonary arteries was performed to rule out the initial suspicion of pulmonary thromboembolism (Figure 1), targeting no thrombi, but revealing marked pulmonary artery dilation, suggestive of significant PH. Given the poor response to diuretics, the patient was evaluated by the Cardiology Department at our center. Transthoracic echocardiography (TTE) (Figure 2) showed marked right ventricular (RV) enlargement and dysfunction, with an estimated pulmonary artery systolic pressure (PASP) of 120 mmHg, suggestive of severe PH.

Fig. 1 Computed angiotomography of the pulmonary arteries. Axial planes at the level of ventricular cardiac chambers and at the level of the great vessels outflow tracts and bifurcation of the pulmonary artery (B). No intravascular filling defects in the pulmonary artery or images suggestive of thrombus (B) are observed. RV enlargement is of note, with a right / left ventricle ratio >1 (A). RV: right ventricle.
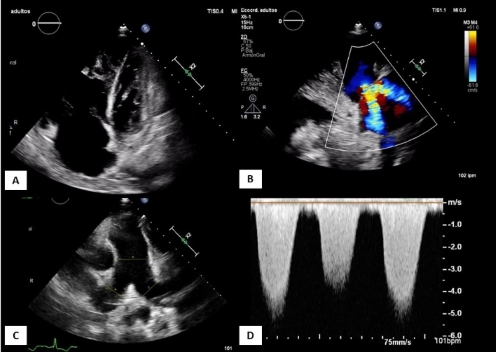
Fig. 2 Transthoracic echocardiography on admission. Apical 4-chamber planes (A) modified parasternal long axis over RV (B), parasternal short axis at the level of great vessels (C) and continuous Doppler over tricuspid regurgitation flow (D). Figure (A) shows severe RV enlargement and dysfunction, with severe functional tricuspid regurgitation (B). Enlargement of the pulmonary artery. The RV-RA gradient of tricuspid regurgitation by Doppler is 120 mmHg (D), suggestive of severe PH. PH: pulmonary hypertension. RA: right atrium. RV: right ventricle
Due to rapid clinical worsening -mainly breathlessness- with increased tachypnea and signs of heart failure, the patient was referred to the Coronary Care Unit to start intravenous therapy with higher doses of dobutamine and furosemide (1 g in continuous infusion). Evacuative paracentesis for symptomatic management of ascites was performed, and Doppler ultrasound confirmed proper functioning of the liver graft. Differential diagnosis was broad, considering that the patient had a history of cirrhosis, dyspnea, and acute heart failure.
Portopulmonary hypertension (PoPH) syndrome is included within group 1 PH, 1 and accounts for 10% of the total cases of this entity. PoPH is defined as pulmonary arterial hypertension (PAH) associated with portal hypertension. 2 It occurs in 1-2% of these patients; 1 in fact, in the REVEAL Registry -a multicenter, observational study on 3000 PAH patients-, the prevalence of PoPH was 5%, more common in women and in autoimmune cirrhosis. 3 Severity is determined by mean pulmonary pressure (mPAP) value; therefore, it is defined as mild PoPH in patients with mPAP < 35 mmHg, and as severe PoPH in cases of mPAP > 45 mmHg. The pathophysiology of PoPH is not known; however, the theory with the most significant impact states that it is due to an imbalance of vasoconstrictor mediators and vasodilators. While most patients are asymptomatic, dyspnea on exertion is the most common symptom, as was the case in our patient. Current European guidelines 1 recommend starting the diagnostic evaluation with TTE, so that, in patients with indirect data of PH (peak tricuspid regurgitation velocity > 2.8 m/s, RV enlargement, pulmonary artery or inferior vena cava enlargement) or with other risk factors, right heart catheterization would be indicated, as was the case in our patient. The study revealed severe precapillary PAH: mPAP 57 mmHg, wedge pressure 11 mmHg, elevated transpulmonary gradient 46 mmHg, and pulmonary vascularresistance (PVR) 14.5 Wood units (WU).
Current hemodynamic diagnostic criteria for PAH include: mPAP > 20 mmHg at rest, wedge pressure 15 mmHg, and PVR > 2 WU. 1
The ultimate criterion for diagnostic certainty of PoPH would be to confirm portal hypertension by clinical signs; in case of doubt, venous catheterization to measure the hepatic venous pressure gradient is suggested. 4
As this criterion was unmet in our patient, diagnosis was "probable portopulmonary syndrome", although isolated cases of de novo diagnosis of PoPH within the first 6 months following transplantation have been described in the literature.
It is an entity with poor prognosis: in the absence of therapy, PoPH has been associated with a 5-year survival rate of 14%; 2 however, a 51% improvement in 5-year survival rate under medical treatment and 81% with liver transplantation have been reported. 5 While positive outcomes with the medical treatment of PAH have been reported, most studies are not aimed at PoPH patients (except for the PORTICO trial, which showed positive hemodynamic outcomes with macitentan, or the PATENT-1 trial, 6 which included a small PoPH population under riociguat treatment, with positive functional outcomes); in any case, this therapy has shown positive hemodynamic and functional effects, but no effect on survival rates. Current guidelines recommend triple combination therapy of endothelin receptor antagonist, phosphodiesterase-5 inhibitor and prostacyclin receptor agonist (Class IIa recommendation) in high-risk patients, as was the case reported here. 1 Our patient was started on epoprostenol, sildenafil and macitentan, showing a slow but progressive improvement, followed by withdrawal of inotropes.
Typically, liver transplantation is contraindicated in these patients due to high perioperative morbidity and mortality, and would only be recommended in patients with liver disease per se requiring transplantation; it was not discussed in the case of our patient since his graft function was normal. Guidelines recommend initiation of PAH therapies in patients with mPAP > 35 mmHg candidates for transplantation; 3 however, it is contraindicated in severe PoPH not improving with medical treatment, since perioperative mortality in patients with mPAP > 45 mmHg is close to 100%.
One month after admission, the patient was discharged on triple therapy and home hospitalization. Follow-up TTE at 6 months showed normal RV function; PAH medication was progressively withdrawn.
Differential diagnosis should consider PH associated to high pulmonary blood flow (with normal PVR and no need to initiate a specific therapy) and hepatopulmonary syndrome (typically presenting without PH, characterized by arteriovenous shunts in the pulmonary circulation, and causing hypoxemia, orthodeoxia and platypnea; liver transplantation is the treatment of choice in severe cases).
Less common entities -which should be considered in patients with a history of liver disease- include cirrhotic cardiomyopathy, other typical causes of heart failure, and extracardiac causes of dyspnea -common in this patient profile- such as anemia, ascites, or hydrothorax.
In conclusion, dyspnea in patients with a history of liver disease has been a challenge for cardiologists, not only because of the wide range of diagnoses to be assessed, but also because of its complex hemodynamic profile. Portopulmonary syndrome is a rare entity. While its standard definition refers to patients with portal hypertension, this entity is currently being described in already transplanted patients -particularly early, in the first 6 months post-transplantation- as in the case reported here; therefore, we must always take it into account, given its poor prognosis and the absence of a specific treatment per se.

