










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista argentina de endocrinología y metabolismo
versão On-line ISSN 1851-3034
Rev. argent. endocrinol. metab. vol.52 no.2 Ciudad Autónoma de Buenos Aires jul. 2015
REVISIÓN
Implicancias fisiopatológicas del receptor androgénico. Mutaciones, polimorfismos y patologías asociadas
Pathophysiological Implications of Androgen Receptor. Mutations, Polymorphisms and Pathologic Associations
Levalle OA, Lalosa S.
División Endocrinología, Hospital Carlos G. Durand, Buenos Aires, Argentina
Correspondencia: División Endocrinología, Hospital Carlos G. Durand, - Díaz Vélez 5044, (1405) Ciudad Autónoma de Buenos Aires Telefax: 011 4982-5212 - E-mail: endodurand@fibertel.com.ar
Recibido: 17-09-2014
Aceptado: 18-11-2014
RESUMEN
Los andrógenos son sintetizados y segregados hacia el torrente circulatorio principalmente como testosterona. Estas hormonas afectan diferentes acciones sobre varios tejidos periféricos. Sus efectos biológicos son mediados en las células blanco a través del receptor androgénico (RA). El complejo RA es miembro de la familia de receptores de hormonas esteroideas, que posee un gran número de coreguladores asociados al receptor. Se encuentra en múltiples tejidos y manifiesta cambios a través del desarrollo, el envejecimiento y transformaciones malignas.
Recientemente, la clonación y caracterización de varios coreguladores del RA permitieron efectuar análisis moleculares sobre distintos aspectos de la fisiología y fisiopatología androgénica. Luego de ingresar a la célula blanco y antes de ejercer su función específica, la testosterona puede ser metabolizada por las aromatasas a estradiol en hipotálamo, hueso y muchos otros tejidos, o alternativamente ser metabolizada por la 5 a-reductasa a dihidrotestosterona en la mayoría de los órganos reproductivos masculinos.
El RA juega un papel crítico en la función de varios órganos, incluyendo los órganos sexuales primarios y accesorios, el músculo esquelético, la célula muscular lisa vascular, la célula endotelial, el sistema nervioso central, el tejido adiposo y el hueso, por lo cual se convierte en un interesante blanco terapéutico.
Una serie de mutaciones del RA se han vinculado con la amplia variabilidad de las manifestaciones clínicas, desde el Síndrome de Insensibilidad Parcial a los Andrógenos (SIPA) hasta el cuadro de feminización completa, al cáncer de próstata, al síndrome de ovario poliquístico, a la infertilidad masculina, al síndrome de Klinefelter, etc.
Por otro lado, se describió que polimorfismos del tracto glutamina del RA puede reducir la función del receptor y aumentar el riesgo de infertilidad y defectos de la espermatogénesis. Cuando la expansión supera las 40 repeticiones causa una rara enfermedad neuromuscular, espinal y atrofia muscular (enfermedad de Kennedy), la que cursa además con menor virilización, atrofia testicular, producción espermática limitada y en consecuencia infertilidad. Estos datos indican que existe una relación directa entre la longitud de esta variable de la región polimórfica y el defecto de la maduración espermática causada por una menor capacidad funcional del RA. Sin embargo, esta correlación no se puede extrapolar a todos los grupos étnicos ya que existen discrepancias en la literatura según la región geográfica y poblaciones evaluadas.
En esta revisión aportamos una breve visión de la acciones genómicas de los andrógenos y su receptor así como la influencia de varios coreguladores. También analizamos los complejos aspectos de la biología molecular del RA y sus coreguladores que están vinculados a diversas enfermedades.
Rev Argent Endocrinol Metab 52:79-107, 2015
Los autores declaran no poseer conflictos de interés.
Palabras clave: Receptor androgénico; Mutaciones genéticas; Polimorfismos genéticos; Andrógenos; Testosterona.
ABSTRACT
Androgens are synthesized and secreted into the blood stream for circulation mainly as a form of testosterone. These hormones affect a number of diverse responses in a variety of peripheral target tissues. Their biological actions are mediated in the peripheral target cell via the androgen receptor (AR). The AR complex is a ligand bound nuclear receptor, combining general transcriptional complex with a large number of receptor-associated co-regulators. The AR is a member of the steroid hormone receptor family, which is found in a variety of tissues, and changes throughout development, aging, and malignant transformation. Recently, cloning and characterization of several AR coregulators have allowed for cellular and molecular analysis of many different aspects of androgen physiology and pathophysiology.
After entering its target cells and before it can exert its specific function, To will either be metabolized by aromatase into estradiol in the hypothalamus, bone and many other tissues, or be metabolized by 5 a-reductase into dihydrotestosterone in most of the male reproductive organs.
Androgen receptor plays a critical role in the function of several organs including primary and accessory sexual organs, skeletal muscle, vascular smooth muscle cell, endothelial cells, central nervous system, adipose tissue, and bone, which makes it a desirable therapeutic target.
Numerous types of androgen receptor mutations have been linked to a wide range of clinical manifestations, from the partial androgen insensitive syndrome (PAIS) to complete testicular feminization, prostate cancer, polycystic ovary syndrome, male infertility, Klinefelter syndrome, etc.
Besides, polymorphism of the AR glutamine tract has been reported to reduce receptor function and increase the risk of male infertility and defective spermatogenesis. When expansion exceeds 40 repeats, it causes a rare neuromuscular disorder, spinal and bulbar muscular atrophy (Kennedy's disease), which is also associated with decreased virilization, testicular atrophy, reduced sperm production, and infertility. These results indicate a direct relationship between the length of this AR-specific variable polymorphic region and defective sperm maturation caused by decreased functional competence of the AR. However, a correlation can not be established between the variable CAG (polyglutamine)-repeat and the presence of illness in all the different ethnic groups.
In this review, we provide a brief overview of genomic androgens/AR actions, as well as of the regulation of their co-regulators. We also explore several complex aspects of the molecular biology of AR and co-regulators that are related to clinical diseases.
Rev Argent Endocrinol Metab 52:79-107, 2015
No financial conflicts of interest exist.
Key words: Androgen receptor; Gene mutations; Gene polymorphisms; Androgens; Gestosterone.
INTRODUCCIÓN
Los Andrógenos (Ao) forman parte de las hormonas esteroideas y presentan funciones esenciales en el desarrollo y fisiología del cuerpo humano. Es conocida su función en los hombres, donde participan en la diferenciación sexual durante la vida intrauterina (diferenciación de conductos de Wolff, glándulas anexas y genitales externos), desarrollo de caracteres sexuales secundarios en la pubertad y pospubertad, regulación del eje gonadal (“feedback” negativo a la LH), inicio y mantenimiento de la espermatogénesis y desarrollo de la masa muscular. En la actualidad se han estudiado y determinado otras acciones importantes aparte del desarrollo sexual, como lo son promover el desarrollo de la masa muscular, regular el comportamiento humano, agresividad, libido, tono vascular, perfil lipídico, masa magra, sensibilidad insulínica y estado trombótico, entre otras acciones no clásica de los Ao(1-6).
Menos conocida pero también de gran importancia es la acción de los Ao en las mujeres, producidos por las células de la teca y precursores de la síntesis de estrógenos por las células de la granulosa. Claras son las acciones fisiopatológicas de los Ao y su receptor en patologías como síndrome de ovario poliquístico, hirsutismo idiopático, acné, etc. Pero poco definidas y conocidas son las acciones fisiológicas de los Ao en la mujer. Recientes estudios mostraron que ratas hembras “knockout” para el receptor de andrógenos (RA) en el ovario, presentaron alteración en su fertilidad, revelando así un rol fisiológico de los Ao en la reproducción femenina, más allá de ser un simple precursor de estrógenos(7). También investigaciones actuales mostraron su importancia fisiológica en la posmenopausia, su acción en la distribución de la grasa, fuerza muscular, estado metabólico e insulinosensibilidad. Algunas de ellas destacan el rol de la dehidroepiandrosterona (DHEA) en la mejoría de la sensibilidad insulínica a nivel muscular, aunque los resultados son controvertidos. Esto podría deberse en parte a que se toman mediciones de DHEA-S en suero y se lo compara con la sensibilidad insulínica, cuando la forma activa es la DHEA no sulfatada. Por otra parte, otras publicaciones hacen referencia a que la DHEA podría ejercer su acción sobre el receptor androgénico (RA) como receptor estrogénico (RE) dependiendo de las concentraciones y características tisulares(8).
Los Ao principales en la mujeres en condiciones normales son la androstenediona, testosterona (To), DHEAS y DHEA. En el hombre los principales son la testosterona y la dihidrotestosterona (DHT). Todos ellos ejercen su acción a través del mismo receptor -el RA-, pero es la afinidad al mismo y la estabilidad del complejo Ao-RA lo que determina la potencia de cada uno de ellos. Por lo anteriormente dicho, la DHT constituye el Ao más potente.
Hablar de implicancias fisiopatológicas del RA nos obliga en primer lugar a hacer una breve reseña de la estructura del receptor, genética y molecularmente, como así también de su mecanismo de acción, para después sí poder pasar a explicar su falla y desarrollo de patologías características. Por último, nos explayaremos en algunas patologías ejemplificadoras de la pérdida y ganancia de función del RA, como son el síndrome de insensibilidad a los andrógenos, el cáncer de próstata, la infertilidad masculina, fenotipos del síndrome de Klinefelter y el síndrome de ovario poliquístico (SOP).
CARACTERÍSTICAS GENÉTICAS Y MOLECULARES DEL RA
El RA constituye una proteína de 110kDa, que forma parte de la súper familia de los Receptores Nucleares. En esta familia encontramos seis subgrupos de receptores (receptor de estrógenos, de hormonas adrenales, de hormonas tiroideas, de andrógenos, del ácido retinoico, y de vitamina D), de los cuales el RA forma parte del subgrupo 4(9). Se encuentra codificado en el brazo largo del cromosoma X (Xq11-12). El gen está constituido por 8 exones que codifican una proteína de 919 aminoácidos. El exón 1 presenta dos repeticiones polimórficas (CAG y GGN) que codifican segmentos variables de poliglutamina y poliglicinas respectivamente (ver Figura 1), ubicadas en el extremo amino terminal que constituye el dominio de transactivación independiente del ligando del receptor (ominio amino terminal: DNT). Estas dos regiones polimórficas están separadas por una secuencia no polimórfica de 248 aminoácidos(10-12).
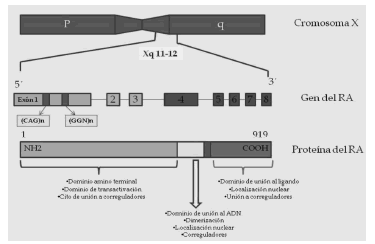
Figura 1. Esquema de la organización cromosómica, genética y proteica del receptor androgénico (RA). Adaptado de Brinkmann A. 2001(12)
CAG es una repetición de longitud variable (entre 8-35 repeticiones) y su longitud se correlaciona inversamente con la actividad del RA(13-15). GGN es una repetición compleja de (GGT)3 GGG(GGT)2(GGC)n que varían en una longitud de 10 a 30 repeticiones. Las repeticiones GGN se han estudiado con menor frecuencia debido a los problemas técnicos que existen en la amplificación de las regiones ricas en GGC (glicina) que están presentes en este polimorfismo(13-15). Si bien, son varias las proteínas que se postularon como interacción de dicho dominio de transactivación, aún no se determinó cuál es proteína correguladora específica para cada tejido y su mecanismo de acción, dado que probablemente haya correguladores diferentes, específicos y con acciones distintas según el tejido que se evalúe(16).
La estructura general del receptor es similar a la de cualquier otro tipo de receptores nucleares. Empezando por el extremo Amino terminal (NH2), ya descripto, donde se encuentra el dominio de transactivación independiente del ligando (DNT) y los también antes nombrados polimorfismos. Un dominio central de unión al ADN (DBD), otro dominio de unión al ligando (LBD) en la región Carboxiterminal (COOH) y finalmente una región “bisagra” que une el LBD con el DBD (4,6,17-20) (Figura 1).
La estructura cristalográfica 3-D del receptor muestra que el sitio de unión al ligando está conformado por doce a hélices y cuatroß dispuestas en dos hojasß, que forman un bolsillo hidrofóbico para el ligando(18). Las doce hélices se doblan en un sándwich de tres capas, las hélices H1/2, H3, H7 y H10/11 forman las dos capas exteriores que unen el ligando mientras que las capas interiores (H4/5, H8 y H9) que no unen el ligando constituyen un corazón hidrofóbico. Este sitio, además de la unión al ligando, participa en la dimerización del receptor y también contiene un dominio de transactivación dependiente del ligando (AF2)(4,6,18,19). A diferencia de otros receptores nucleares, el RA posee uno o más dominios de transactivación; el dominio AF1 se caracteriza por una activación independiente del ligando. Tal es la importancia de dicha región que colocado el RA en presencia solo de Ao, sin ninguna otra sustancia, mostró una actividad débil. Esto determina que las acciones del RA están sujetas a una modulación positiva o negativa por un número, aún no dilucidados, de correguladores(13,15). El NTD interactúa fuertemente con el LBD (interacción NH2-COOH), de manera dependiente de la hormona; esto es importante para poder interaccionar con los coactivadores y así regular la transcripción(13,15-17,20,21).
MECANISMO DE ACCIÓN DEL RA
El RA en ausencia de Ao se encuentra inactivo en el citoplasma, unido a proteínas de Shock térmico (Hsp70 y Hsp90) y sus chaperonas formando un complejo dinámico. En presencia de Ao se produce la disociación del receptor a las Hsp y la unión al Ao, con posterior dimerización del receptor, formando homodímeros, traslocación al núcleo y unión al ADN en los elementos de respuesta a la hormona, para facilitar la transcripción o no de los genes regulados por los Ao. El dominio NH2 terminal constituye más del 60 % de la proteína que conforma el RA y contiene una región de regulación de la actividad del receptor, al unirse a coactivadores y correpresores específicos de tejidos, que determinan así la especificidad y selectividad de la acción hormonal en el tejido, junto a los promotores de cada gen. El dominio de unión al ligando consta de 20 residuos de aminoácidos que interactúan más o menos directamente con el Ao(22) (Figura 2).
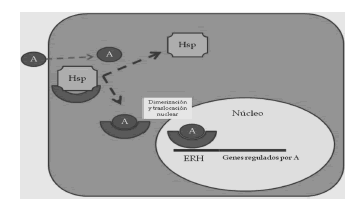
Figura 2. Mecanismo acción del RA. Los andrógenos difunden a la célula y se unen al RA en el citoplasma, se dimeriza receptor y activado se transloca al núcleo, donde ocupa los elementos de respuesta a la hormona (ERH) en las regiones promotoras de los genes regulados por andrógenos, estimulando o bloqueando la transcripción proteica. Tomado y modificado de Brinkmann A(19)
FISIOPATOLOGÍA DEL RA. MUTACIONES Y POLIMORFISMOS
Cabe recordar, cómo se nombró en la introducción, la importancia de los andrógenos en niveles séricos fisiológicos no solo en hombres sino también en mujeres, para el normal desarrollo de caracteres sexuales y reproductivos, como así también el buen desarrollo de vías metabólicas.
Las vías de los Ao se integran a otras vías que regulan varios procesos metabólicos en el cuerpo humano, esto determina que una alteración en la acción de los andrógenos no solo se ve reflejada en alteraciones en la diferenciación o mantenimiento de los caracteres sexuales, sino también en varios otros órganos y procesos metabólicos, circulatorios, etc. Es por eso que la vía de los Ao ha sido estudiada en un número importante de patologías más allá de las alteraciones en la virilización o trastornos reproductivos de los pacientes(1-6).
Las dos formas de compromiso de la acción del RA a la que nos abocaremos en esta revisión son las mutaciones y polimorfismos de su gen. Polimorfismo es una variación frecuente en la secuencia de bases de un lugar determinado del ADN entre los individuos de una población, presentes en más del 1 % de dicha población. En tanto una mutación son cambios pocos frecuentes en las secuencias de bases del ADN, aparece en menos del 1 % de la población. Se han descripto diferentes puntos de mutaciones, deleciones o inserciones a nivel del gen del RA que modifican la actividad y que son las responsables del desarrollo de enfermedades como el síndrome de insensiblidad completa a los andrógenos (SICA), la insensibilidad parcial a los andrógenos (Síndrome de Reifenstein o SIPA), el cáncer de próstata (CP), etc. (23,24)
Por otra parte se ha visto que la actividad del receptor está influenciada por un polirmofismo genético del exón 1 de dicho receptor, donde se hallan dos secuencias, ya nombradas, de repeticiones polimórficas de número variable, CAGn y GGNn, que codifican una poliglutamina y poliglicina respectivamente. Aunque las consecuencias en la longitud del fragmento GGN no son claros, varios estudios in vitro han puesto en evidencia una relación inversa entre el número de repeticiones CAG (normal de 8-35) y la actividad transcripcional del RA. Los hombres con una longitud CAG superior a 28 repeticiones tienen una mayor incidencia de deterioro de la espermatogénesis e infertilidad, mientras que una longitud superior a 40 se relacionó con el síndrome de atrofia muscular bulbar y espinal (SBMA), un raro trastorno neuromuscular caracterizado por atrofia muscular espinal y bulbar asociado a una insensibilidad a los andrógenos leve, que describiremos más adelante. A la inversa, un número de repeticiones cortas de poliglutaminas determinan un RA transcripcionalmente más activo y fue relacionado al cáncer de próstata, pubertad prematura, calvicie temprana masculina, hirsutismo idiopático en la mujer, diferentes fenotipos del Síndrome de Klinefelter y SOP entre otros, muchos de ellos con resultados contradictorios(25-32).
Las repeticiones GGN se han estudiado con menor frecuencia debido a los problemas técnicos que existe en la amplificación de las regiones ricas en GC que están presentes en este polimorfismo(13).
Es por lo dicho hasta aquí que en esta revisión se hará foco en mutaciones del RA determinante de patologías, de las cuales se tomará como ejemplo la insensibilidad a los andrógenos (con todo el espectro fenotípico que conlleva según el tipo de mutación); como así también patologías relacionadas con las variantes polimórficas de CAG, de las cuales nombraremos el cáncer de próstata, la infertilidad masculina, fenotipos de Klinefelter y el SOP en la mujer.
SÍNDROMES DE INSENSIBILIDAD A LOS ANDRÓGENOS (SIA)
A) Características Generales del SIA
El SIA forma parte de los llamados Trastornos del Desarrollo Sexual, denominación utilizada para nombrar a todas aquellas alteraciones congénitas del desarrollo en que el sexo cromosómico, gonadal o anatómico son atípicos, es decir trastornos que originan un sexo fenotípico diferente al del genotipo. Esta nueva denominación originada en el consenso de Chicago se ha creado para evitar el uso de nomenclaturas estigmatizantes para el paciente y sus familias. Así, el pseudohermafroditismo masculino y femenino se denominan trastornos del desarrollo XY y XX respectivamente y el hermafroditismo verdadero se denomina trastorno del desarrollo ovotesticular. Este consenso clasifica las alteraciones del desarrollo sexual según el cariotipo en 3 categorías: a) Alteraciones de los cromosomas sexuales, b) Alteraciones del desarrollo 46,XX, c) Alteraciones del desarrollo 46,XY. Es en este último grupo que encontramos el SIA(33-35)(ver Tabla 1).
TABLA 1. Clasificación de las alteraciones del desarrollo sexual

La diferenciación sexual masculina ocurre cuando un cigoto XY dirige la diferenciación a masculino, es decir la presencia del gen SRY (en el brazo corto del cromosoma Y), determina la aparición de los testículos, los cuales a su vez produciendo Ao y hormona antimülleriana (AMH) dan origen a genitales externos e internos masculinos. La falta de Ao o su imposibilidad de actuar, lleva a la diferenciación sexual genital y fenotípica femenina. Es decir que en el SICA la falta de acción de los Ao, por mutación del RA del cual los hombres solo tienen una copia, origina un fenotipo femenino aunque sea cromosómicamente XY36 (ver Figura 3).
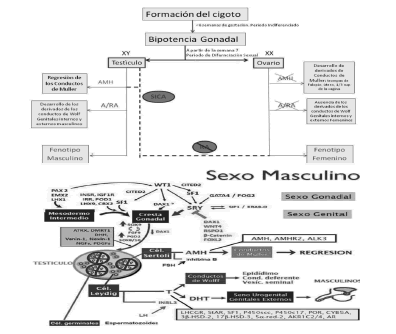
Figura 3. Arriba: Esquema simplificado de la diferenciación sexual. RA: receptor androgénico. SICA: insensibilidad completa a los Ao. AMH: hormona antimülleriana. Abajo: Esquema que muestra la compleja expresión génica que lleva a la diferenciación sexual. Tomada de Leuan A y col.(37).
Las mutaciones con pérdida de función del RA abarca un amplio grupo de características fenotípicas de insensibilidad androgénica que van desde un fenotipo femenino completo (Síndrome de Insensibilidad Completo a los Andrógenos, SICA) pasando por mutaciones menos severas que originan trastorno en la virilización (Síndrome de Insensibilidad Parcial a los Andrógenos, SIPA) u origen solo de infertilidad aislada por alteraciones espermáticas como única manifestación de la alteración del RA (Síndrome de insensibilidad Mínima a los Andrógenos, SIMA) (ver Figura 4).

Figura 4. Diferentes grados de insensibilidad a los andrógenos, abarcando desde el más severo, que genera una insensibilidad completa y fenotipo femenino (SICA), hasta un grado mínimo con solo infertilidad (SIMA o SIPA1). En el medio, los diferentes grados de insensibilidad parcial (SIPA). Modificado de Baculescu N. (38).
B) Clínica del Síndrome de Insensibilidad completa a los andrógenos (SICA)
John Morris fue el que por primera vez describió este síndrome, al revisar las características de 82 pacientes que eran niñas y mujeres con testículos que parecían producir una hormona similar a los estrógenos, y los llamó “testículo feminizante”. Dado lo contradictorio y estigmatizante de ese nombre, después de estudios que permitieron comprobar que la verdadera causa del desarrollo de un fenotipo femenino en pacientes XY, era la falta de acción androgénica, se lo renombró “Síndrome de Insensibilidad a los Andrógenos”. Como se mencionó anteriormente los diferentes tipos de mutaciones originan distintos grados de insensibilidad androgénica y por ende de presentación clínica-fenotípica(39,40). Las formas frecuentes de presentación del SICA suele ser por amenorrea primaria en una adolescente con desarrollo puberal normal, o por distención abdominal y/o hernia inguinal en un bebé o en la infancia. Lo más común es una niña con desarrollo puberal normal, con crecimiento de tejido mamario pero ausencia de sangrado menstrual. La presencia normal de caracteres sexuales secundarios dependiente de estrógenos se produce dado que el exceso de andrógenos circulantes (en rango masculino) se transforman en estrógenos por aromatización periférica(41-43). El vello axilar y pubiano (dependiente de Ao Suprarrenales) suele estar ausente o ser muy escaso. En la infancia el SICA se puede presentar como una hernia inguinal o tumefacción labial, que contiene un testículo, en una niña/bebé aparentemente femenino. Las hernias inguinales bilaterales en un recién nacido femenino son raras, pero alcanza un 1-2 % de los SICA durante la infancia(42,43); por eso se recomienda la realización de un cariotipo y/o una biopsia de la gónada que está dentro del saco herniario(42). Una alternativa puede ser medir la longitud vaginal en las niñas prepúberes sometidas a una cirugía por hernia inguinal, así una vagina corta con ausencia de útero y trompas de Falopio obliga a la realización de un cariotipo. Esto se debe a que la presencia de AMH producida por las células de Sertoli testiculares, lleva a una regresión y consecuente ausencia de todos los derivados de los conductos de Müller, como son las trompas de Falopio, el útero, el cuello uterino y la vagina proximal(36). La vagina en el SICA varía desde un hoyuelo en el periné a una longitud casi normal, pero siempre se caracteriza por terminar en un fondo de saco ciego.
En la actualidad el sexo fetal es cada vez más conocido antes del nacimiento a través de cariotipos realizados en biopsia de vellosidades coriónicas, análisis de líquido amniótico en el que se busca ADN fetal libre, como así también la ecografía en 3D. Por lo tanto el SICA debería sospecharse y buscarse cuando el sexo prenatal no coincide con el fenotipo del recién nacido(45,46).
Otra forma de presentación y diagnóstico de SICA, es el antecedente familiar de SICA con una transmisión ligada al X, en ocasiones también puede ser un caso aislado sin antecedentes familiares(47).
Las niñas con SICA presentan un desarrollo puberal a la edad adecuada, no se detectó una pubertad retrasada en niñas con SICA comparada con niñas sin síndrome. Las niñas y mujeres adultas con SICA poseen un nivel de Ao muy superior al de las mujeres sanas. Estos niveles son superiores en pacientes en los que la gonadectomía se retrasa(48,49). La mayor estatura final en mujeres con SICA se debe a la presencia de la región reguladora de la talla en el brazo largo del cromosoma Y, aunque existen varios loci en todo el genoma determinantes de la talla final. El hecho de que mujeres con SICA tengan talla similar a la masculina, hace referencia a la importancia de la presencia del cromosoma Y en la talla, más que la exposición a los Ao(50-52).
C) Clínica del Síndrome de Insensibilidad Parcial a los Andrógenos (SIPA)
La clínica de presentación del SIPA (antes también llamado Síndrome de Reifenstein) depende del grado de respuesta a los andrógenos por parte de los genitales externos e internos. Así se pueden hallar desde un fenotipo predominantemente femenino (con clitoromegalia, hasta fusión de labios), genitales ambiguos, o un predominio de fenotipo masculino (con micropene, hipospadias perineal, criptorquidia). La presencia de derivados de los conductos de Wolff depende de la bioquímica y fenotipo del SIPA(37,38).
Los diagnósticos diferenciales con el SIPA, se detallan en la Tabla 2.
TABLA 2. Diagnósticos diferenciales del Síndrome de Insensibilidad Parcial a los Andrógenos

Se han realizado guías para el estudio y diagnóstico de SIPA y otros trastornos del desarrollo sexual, tanto en recién nacidos como en adolescentes, pero no son el objetivo de esta revisión. Solo cabe señalar que para confirmar el diagnóstico de SIPA sería necesaria la secuenciación genética del RA. De ser posible se puede realizar una prueba funcional in vitro que confirme la menor función del receptor. Ya que, por ejemplo, la hipospadia es la malformación masculina más frecuente (más de 8 casos por 1000 nacidos vivos) asociado a bajo peso al nacer y se desconoce la causa(53,54).
D) Clínica de Síndrome de Insensibilidad Mínima a los Andrógenos (SIMA)
Es producido por una mutación en el RA infrecuentemente reportada, en general se presenta como un hombre con alteración en su fertilidad sin malformaciones genitales. Se ha demostrado en ratones “knockout” para RA, lo esencial que es la expresión del mismo en las células de Sertoli y Leydig y como lo es también la intercomunicación entre estas células para una adecuada espermatogénesis. El índice LH/To sérica puede ser usado como indicador de SIMA en pacientes con infertilidad y se podría utilizar como método de screening para buscar la mutación del RA en estos pacientes. Es posible, si la infertilidad solo se debe a una disminución en el conteo espermático, que pueda resolverse con el tratamiento con análogos de testosterona (To), para vencer la resistencia androgénica(55-57). El SIMA también puede hallarse en el contexto de una patología sistémica como lo es la Atrofia Muscular Bulbar y Espinal, cuya causa de disfunción del RA es un polimorfismo del RA en el exón 1 y no de una mutación, del cual nos ocuparemos más adelante(58).
E) Características endocrinológicas del SIA
Los pacientes con SIA se caracterizan por tener gónadas con producción hormonal intacta lo que lleva a un alto nivel de To (en rango normal para niños u hombres de igual edad), con LH en niveles inapropiadamente elevados. Dado que la producción y acción de las hormonas generadas por las células de Sertoli se encuentran conservadas, los niveles de Inhibina B y por ende los de FSH son normales. El exceso de To es convertido en estrógenos por aromatización periférica, que sumado a la producción directa testicular inducidos por el aumento de LH, genera niveles de estrógenos considerablemente mayores a los hallados en un hombre, pero menores que en una mujer normal. Los altos niveles de LH pregonadectomía, y el mayor aumento de LH y FSH posgonadectomía, son parcialmente suprimidas por la terapia estrogénica. Esto indica la presencia de una resistencia hipotálamo-hipofisaria al “feedback” negativo de los Ao, que explica los niveles altos de LH pregonadectomía, pero persistencia de respuesta a la regulación estrogénica. La SHBGen condiciones normales presenta un dimorfismo sexual, así los andrógenos disminuyen su producción hepática en tanto los estrógenos la aumentan. Por todo lo dicho los pacientes con SIA presentan niveles de SHBG similares a los de una mujer, sin diminuir tras la administración de Ao(59-61)(ver Figura 5).

Figura 5. Esquema simplificado de las características endocrinológicas en el Síndrome de Insensibilidad a los Andrógenos. To: testosterona, LH: hormona leutinizante, FSH: hormona folículoestimulante, Eo: estrógenos, AMH: Hormona antimülleriana, SHBG: globulina transportadora de hormonas esteroideas
El patrón de altos niveles de To con elevación de gonadotrofinas es característico de SIA y debería sospecharse al hallarlo en un niño. Durante los primeros meses de vida la LH induce un aumento de To, hecho de importancia fisiológica llamado “mini pubertad” del neonato. Esto ocurre en el desarrollo normal y es lo que imposibilita utilizar el aumento de gonadotrofinas y To como patrón de sospecha diagnóstica del SIA en los primeros meses de vida. Pero el índice elevado de LH/To es de utilidad en la infancia, período de “quiescencia gonadal”, donde en niños sanos los valores de To y LH se mantienen bajos. En el SIMA dicho índice es el parámetro de mayor utilidad en caso de sospecha, para la realización de “screening” de mutación del RA(62-65). Otra diferencia característica entre los niños sanos y con SIA, es que estos últimos suelen tener una hiperrespuesta de la To al estímulo con gonadotrofina coriónica humana (HCG), sean SICA o SIPA. Por lo tanto el test de HCG es útil para hacer un diagnóstico diferencial inicial con otras causas que pueden originar un fenotipo similar al SIPA(62-65).
La AMHes producida por las células de Sertoli en el hombre y en mucho menor medida por las células de la granulosa en la mujer pospuberal. Los niveles de AMH en condiciones normales también se elevan en período neonatal y presentan un dimorfismo sexual (valores más elevados en los hombres que las mujeres). También, en condiciones normales los Ao inhiben en Sertoli la secreción de AMH, pero esto no ocurre en los primeros años de vida ya que estas células carecen de RA. Estos receptores se van adquiriendo en la infancia y alcanzan un número considerable en la pubertad, determinando en ese momento la caída de los niveles de AMH, que alcanzan una inhibición marcada al llegar al estadio III de Tanner. Por lo dicho, los niveles de AMH y To se comportan inversamente durante la pubertad, pero esto no se respeta en la mini pubertad del neonato, dada la ausencia de RA en la célula de Sertoli en los primeros años de vida, lo que sugiere un estado de insensibilidad transitoria y fisiológica de dicha célula durante la vida fetal y neonatal de desarrollo testicular. Esta insensibilidad se mantiene y prolonga en el SIA, por la falta de acción del RA, y esto explica porque los dosajes de AMH son mayores en niños con SIA que en los normales y persisten elevados incluso en la pospubertad y adultez, si no se realiza gonadectomía. Los altos niveles de AMH permiten diferenciar el SIA de otros síndromes de disgenesia gonadal(66-69)(ver Figura 6).
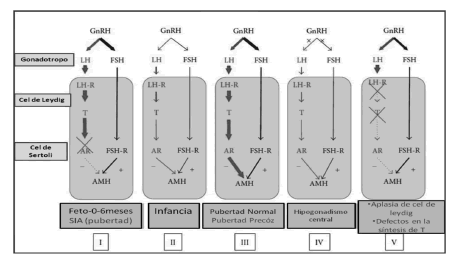
Figura 6. Regulación de la secreción de AMH testicular por gonadotrofinas y andrógenos. GnRH estimula la secreción de LH y FSH. LH actúa sobre su receptor (LH-R) en células de Leydig, induciendo la secreción de testosterona (To). FSH actúa sobre el receptor de FSH (FSH-R) en células de Sertoli. El eje hipotálamo-hipófiso-gonadal está activo en el feto y primera infancia, en reposo durante la niñez y se reactiva en la pubertad. La FSH es un inductor moderado de la secreción de AMH, mientras que la To, actuando a través del receptor de andrógenos (AR), es un potente inhibidor de su producción. En el feto normal, en la infancia y en pacientes con el síndrome de insensibilidad a los andrógenos (SIA), la falta de la expresión/acción del AR resulta en una alta producción de AMH por las células de Sertoli (I). Durante la niñez hay un estado hipogonadotrófico fisiológico que resulta en muy baja To y altos niveles de AMH, pero algo más bajo probablemente debido a la falta de estímulo de FSH (II). En la pubertad normal o precoz la To prevalece sobre la FSH, lo que resulta en la inhibición de AMH (III). En el hipogonadismo central congénito la AMH es menor que en el niño normal debido a la larga ausencia de estímulo de FSH en la vida fetal. Sin embargo, en la pubertad el efecto inhibidor de To está también ausente, por lo que AMH persiste en niveles mayores que en la pubertad normal (IV). En el hipogonadismo primario específico de las células de Leydig (aplasia o hipoplasia de las células de Leydig por defectos en el LH-R o en la esteroidogénesis), el efecto inhibidor de los andrógenos está ausente y los niveles de AMH son altos. La zona naranja representa el testículo. El grosor de las líneas está en correlación con el efecto de hormonas en su objetivo. Tomado y modificado de Grinsponand R, Rey R.(72).
Los diagnósticos diferenciales principales en niñas con amenorrea primaria, más allá del SICA son todas las otras causas de disgenesia gonadal completa, síndrome de Mayer-Rokitansky-Kuster-Hauser, otras anomalías de los derivados de conducto de Müller como por ejemplo septos vaginales transversales y otro diagnóstico diferencial importante como el de trastornos en la biosíntesis de andrógenos(70,71).
F) Patogénesis molecular del SIA
Existen 4 tipos de mutaciones descriptas en pacientes con SIA: A) Mutuaciones puntuales (con sustitución de aminoácidos) B) Inserciones o deleciones de nucleótidos (llevan al cambio del marco de lectura o terminación prematura) C) Deleciones parciales (10 nucleótidos) o completas del gen. D) Mutaciones Silenciosas (mutaciones de intrones en sitios de empalmes, sean en el sitio donante o aceptor que afectaran el corte y empalme del ARN) (72). Mas de 800 mutaciones fueron descriptas para el RA en la bases de datos de Cambridge hasta septiembre de 2011. En el síndrome parcial y completo de insensibilidad a los andrógenos, las mutaciones se extienden a lo largo de los 8 exones, pero con una localización preferencial sobre los dominios de unión al ADN (DBD) y dominio de unión al ligando (LBD), dado la compleja y esencial estructura de dichos dominios, resultando en una alteración de la función del RA(73).
Los fibroblastos de la piel genital expresan el RA a altas concentraciones y se pueden utilizar para identificar mutaciones tras la toma de una biopsia de piel. En el SICA hay una ausencia de unión al receptor, en tanto el SIPA y SIMA se altera la constante de afinidad de unión (Kd) al mismo, aunque también en cualquier SIA puede haber una unión Ao-RA conservada con solo la alteración en la actividad de dicho complejo. Los fibroblastos se pueden utilizar para evaluar mutaciones que alteran el corte y empalme del ARNm del RA, cuantificar la expresión del RA por western blots, como así también el estudio de la variación fenotípica registrado en pacientes con la misma mutación(75,76). Con la secuenciación del gen del RA es posible identificar una mutación en más del 95 % de los casos de SICA. La actividad transcripcional del receptor debería ser evaluada en las mutaciones que dan lugar a un fenotipo de SIPA y SIMA, sobre todo en aquellos pacientes en los que se les asignó un sexo masculino, en los cuales dichos ensayos tendrían una importancia en el posible tratamiento hormonal del paciente(77,78). La severidad de la mutación y función del RA se correlaciona con el grado de diferenciación y virilización del feto en el útero. Así mutaciones severas con actividad prácticamente nula del RA se presentan con un fenotipo femenino característico del SICA76. Las mutaciones espontáneas fueron halladas en el 30 % de los casos de SICA, coincidente con otras enfermedades recesivas ligadas al X, como lo es la Hemofilia A por déficit del factor VIII y como la Distrofia Muscular de Duchenne, entre otras(80). La mayoría de las mutaciones de novo ocurren en postcigota, originando un mosaicismo somático del RA mutante, con alta variabilidad fenotípica. También puede existir una variación fenotípica para familiares afectados que heredan la misma mutación(81-83).
Como ya se dijo la mayoría de las mutaciones del receptor se encuentran en el dominio de unión al ligando (LBD), siendo el defecto funcional más común la alteración en el bolsillo hidrofóbico de unión al ligando, lo que conlleva a una dificultad en el reposicionamiento de las 12 a hélices, que permite la activación del receptor y la interacción con el dominio AF-2 y sus correguladores correspondientes(81). Tanto en el SIA como en el cáncer de próstata se han encontrado mutaciones en 14 de los 20 aminoácidos que forman el bolsillo hidrofóbico, que determinan una alteración en su estructura cristalográfica, que explicaría su alteración funcional(72).
La mayoría de las mutaciones localizadas en el LBD entre los residuos de aminoácidos 688 y 712, 739 y 784, y 827 y 870 causan un SICA. Los dos primeros de estos grupos constituyen la mayor parte del sitio de unión al ligando. Una mutación en la 5ta. hélice en el residuo aminoacídico 752 que cambia una arginina por una glutamina (R752Q), lleva a una pérdida del puente de hidrógeno que une la cetona del anillo A del andrógeno. Esta mutación se traduce en un SICA(84,85). La mutación más común reportada de la región C-terminal del RA (H917R) es asociado a un SICA(86,87).
En cuanto a las mutaciones del dominio de unión al ADN (DBD), se han descripto en total 32 mutaciones; 15 en el primer dedo de Zinc y 17 en el 2do dedo de Zinc(70). En este caso las mutaciones permiten la unión correcta entre el andrógeno y su receptor pero hay un defecto en la dimerización y/o unión al ADN y consecuentemente alteración en la transcripción(88,89).
Una observación interesante fue la mutación encontrada en el segundo dedo de Zinc, donde uno o dos de los residuos de Arginina adyacentes (Arg607 y Arg608) se encontraba mutado en pacientes con SIPA que presentaban cáncer de mama. Se postuló que una pérdida de la acción de los andrógenos en las células de la mama predispone a su desarrollo, por la falta de su efecto protector en la mama. Pero falta información para determinar si la mutación del RA tendría una implicancia en la activación de genes estrógenos dependientes(90).
Cualquier mutación en los residuos de cisteína llevaría a una alteración en la estructura de los dedos de zinc, característicos y esenciales para la función adecuada del receptor, llevando a un SIA a pesar de que la unión Ao-RA está conservada. Una de las a hélice en el primer dedo de Zinc se encuentra altamente conservada en todos los receptores de andrógenos, contacta con el ADN e interacciona con pares de bases específicas. Ensayos realizados mostraron que el cambio de la Valina del Codón 582 (del 1er dedo de zinc) por fenilalanina lleva a una falla del RA dando origen a un fenotipo de SICA, en tanto si la sustitución es por Glicina y leucina da origen a un PAIS. El segundo dedo de zinc está constituido por los aminoácidos entre 596-600 y participan en la dimerización del receptor. Mutaciones en esta región son impredecibles. La base de datos de Cambridge registra una mutación en la región C-terminal I604S en gemelos con SICA. Una sustitución del residuo adyacente (D605Y) fue descripto en pacientes con diagnóstico de SIPA que se le asignó sexo masculino, como así también SIPA asignados a sexo femenino(91,92).
El dominio de transactivacion es el más grande (557aa) de los tres dominios y es el menos conservado evolutivamente. Si bien no se ha determinado su estructura cristalográfica, se cree por predicción de análisis bioquímico, que se trata de un dominio desorganizado con regiones intermitentes y estables de a-hélice. Audi y colaboradores(93) reportaron un único caso de un fenotipo de SICA, debido a una mutación de cambio de sentido, situada en dicho dominio. Las mutaciones de cambio de sentido que se hallan en este dominio de transactivación en general se relacionan con fenotipos leves de SIA(94). Los autores concluyen que las mutaciones en el dominio amino terminal, ocurren raramente, y en general son inserciones o deleciones que llevan a la aparición de un codón stop, con la terminación prematura de la transcripción del RA72.
Un grupo raro pero interesante de mutaciones son las halladas en el sitio dador y aceptor de corte y empalme del gen del RA, en pacientes con SIA. En todos los sitios de corte y empalme el sitio dador constituye una secuencia de GTAAG/A, se han reportado 5 mutaciones en el sitio dador, son todas sustituciones, ya sea en posición +1 (G > A or G > T) o en posición +3 (A > T) y resulta en un empalme defectuoso de uno o más exones(72).
TRASTORNOS ESPECÍFICOS ASOCIADOS A POLIMORFISMOS Y MUTACIONES DEL RECEPTOR ANDROGÉNICO
A) Generalidades
Como ya nos hemos referido, los segmentos de polinucleótidos CAG y GGN, en los últimos años han sido de gran interés en el estudio de patologías relacionadas con un aumento o disminución de la actividad androgénica, tales como: la atrofia muscular bulbar espinal (SMBA), el cáncer de próstata, la infertilidad masculina, la calvicie masculina temprana, el hirsutismo idiopático, el SOP, el cáncer de mama, entre otros. Así un amento de número de repeticiones CAG se asoció a una disminución de la actividad del RA y en tanto un corto número de repeticiones a un aumento de actividad del mismo(25-27) (ver Figura 7).

Figura 7. Se muestra gráficamente la relación inversa entre la longitud de CAG y la actividad del RA. Alelos con alto número de repeticiones CAG presentan baja actividad del RA que se expresa como, por ejemplo, en una alteración en la espermatogénesis, mayor riesgo de desarrollo de cáncer de mama, hasta patologías severas y mortales al superar un umbral de números de repeticiones como el SBMA (atrofia muscular bulbar espinal), dónde también existe disminución de la virilización y defectos en la espermatogénesis. Por el contrario, un corto número de repeticiones CAG llevan al aumento de la actividad del RA, facilitando el desarrollo de patologías como la calvicie temprana masculina, hirsutismo en mujeres, como así también aumento del riesgo de presentar cáncer de próstata. Modificado de Yong E. et al.(104).
Ya ha sido probado que el aumento en el número de repeticiones de trinucleótidos es causa de varias enfermedades neuromusculares como el síndrome de X frágil, la enfermedad de Huntington, la ataxia espinocerebelosa tipo 1, atrofia dentatorubral-pallidosluysian, la enfermedad de Machado-Joseph y la ataxia de Friedreich. Se demostró que los pacientes con SBMA tienen un número de repeticiones de CAG en el exón 1 del gen del RA, igual o mayor 40, en comparación con 35 o menos de la población sana. El mecanismo por el cual la expansión de CAG produce este síndrome, se desconoce y es de intensa investigación. Estos pacientes tienen además pruebas de función de RA reducida, a lo cual haremos referencia más adelante(95,96).
El estudio de estos polimorfismos, muestra resultados variables en el número de repeticiones en las diferentes patologías y en las diferentes poblaciones. Este es el primer inconveniente de los estudios y lo que justifica en parte los resultados disímiles cuando se evalúa asociación con las diferentes patologías, así los americanos y los asiáticos tienen un menor número repeticiones de CAG (8-30 y 11-31 repeticiones respectivamente), mientras que los europeos tienen las repeticiones más largas (8-39 repeticiones) en condiciones normales. Además los diseños de los estudios pueden ser también una causa de la variabilidad de resultados, por ejemplo como se seleccionan los casos y los controles, edad media de los mismos, etc.(97,98). Otro hecho que también lleva a una variabilidad es que en los estudios de mujeres hay que considerar el número de repeticiones de los dos alelos(99), tomando clasificaciones de alto-alto o bajo-bajo número de repeticiones; esto se complica aún más cuando se tiene en cuenta la inactivación de uno de los cromosomas X(100).
Para comprender cómo la longitud de repeticiones se relaciona con la actividad del RA es importante también el estudio del medio coregulador del RA, ya que el trinucleótido CAG se encuentra en el dominio de transactivación del receptor. Existen más de 70 proteínas que interactúan con el RA, así es probable que la longitud de repeticiones de CAG interfiera en la relación del RA con su co-regulador(101). Los estudios in vitro han demostrado que la interacción entre el RA y el coactivador ARA24 disminuye con el mayor número de repeticiones CAG, lo que resulta en una disminución de la transactivación del RA(102). Del mismo modo, los alelos del RA con más corto número de repeticiones de CAG son más y mejor coactivados por los miembros de la familia de coreguladores de receptores esteroides (SRC) como el SRC-1, el SRC-3 y el factor intermediario de transcripción 2 (TIF-2) (103). También alternativamente, polimorfismos en las regiones promotoras de los genes diana del RA, alteran la interacción y/o actividad transcripcional, contribuyendo al desarrollo de ciertas patologías.
B) Polimorfismo del RA y el síndrome de atrofia muscular bulbar y espinal (SBMA)
La SBMA también llamada enfermedad de Kennedy, es un desorden caracterizado por una debilidad y atrofia muscular progresiva. Los síntomas generalmente aparecen entre la tercera y quinta década de vida, como resultado de una severa depleción de las motoneuronas inferiores de la médula espinal y bulbar. Compromete a hombres, generando debilidad muscular incapacitante que se inicia en la adultez y empeora con el paso del tiempo. La debilidad en brazos y piernas lleva a una inestabilidad, caídas y menor deambulación. Algunos músculos de la cara y la garganta también se comprometen (músculos bulbares), causando progresivamente trastornos en la deglución y en el habla. También son comunes los tics y fasciculaciones musculares. Se asocia con alteraciones endocrinológicas que incluyen atrofia testicular, infertilidad, ginecomastia, elevados niveles de LH y estradiol. La diferenciación y desarrollo sexual son normales en los estadios iniciales, pero estas alteraciones aparecen con el paso del tiempo. Todos estos trastornos son producidos por un aumento del números de repeticiones de CAG en el exón 1 del RA, variando entre 38-75 repeticiones encontrado en todos los pacientes(95,96,104). De los hallazgos clínicos se puede concluir que el SBMA es resultado de una combinación de alteración de metabolización del RA con aumento de número de repeticiones (que lleva a su acumulación en la motoneurona, originando toxicidad y muerte de la misma) y pérdida de función del RA en los tejidos diana (llevando a un cuadro clínico de insensibilidad a los andrógenos leve). La severidad del cuadro clínico, la edad de presentación de los síntomas y daño de las motoneuronas se correlaciona directamente con el número de repeticiones. Si bien el mecanismo por el cual el RA participa en la muerte neuronal se desconoce, se cree que se debería a que el aumento del número de repeticiones de poliglutaminas lleva al procesamiento aberrante y agregación del receptor, originando la acumulación, toxicidad y muerte neuronal(102). Recientes estudios han mostrado la importancia de la Caspasa 3 (activada por la poliglutamina) en la muerte neuronal(106).
En caso de antecedentes en la familia, el estudio del número de repeticiones CAG se utiliza para el “screening” prenatal de dicha enfermedad. Esta determinación prenatal no solo es útil para detectar los pacientes en riesgo, sino que también es de ayuda para predecir la edad de comienzo de la misma, ya que a mayor número de repeticiones se caracteriza por mayor severidad del cuadro clínico y una edad de presentación más precoz(108).
C) Polimorfismo del RA y cáncer de próstata.
La asociación con el número de repeticiones CAG también se estudió en tumores andrógeno-dependientes como el cáncer de próstata (CP), donde es conocido el uso del bloqueo androgénico como recurso terapéutico. Se evidenció una relación entre los alelos con un corto número de repeticiones y pacientes con CP. La prevalencia de alelos cortos (<22 repeticiones) fue alta (75 %) en hombres afroamericanos con alto riesgo de desarrollar CP, intermedia (62 %) en paciente blancos no hispanos con riesgo intermedio para CP y fue bajo (49 %) en asiáticos con muy bajo riesgo de presentar CP. También se encontró una asociación entre alelos cortos y mayor severidad del CP(109). Los hombres con 18 o menos repeticiones CAG tienen 1,5 veces más riesgo de cáncer de próstata, que aquellos con 26 o más repeticiones(110). Se encontró también que pacientes con menor número de repeticiones presentaban mayor extensión extra prostática y metástasis a distancia. Se halló una disminución del riesgo del 3 % de cáncer de próstata para cada repetición adicional de CAG (111). En conjunto, todos los datos sugieren que las cortas repeticiones de CAG dan lugar a un aumento de la actividad del RA, resultando en una estimulación prostática anormalmente elevada, favoreciendo la presentación del tumor a una edad de inicio más temprana, aumento del grado y severidad tumoral, como así también mayor riesgo de extensión extra prostática.
Varios estudios mostraron asociación entre cáncer de próstata y número de repeticiones CAG, sin embargo otros estudios evidenciaron una ausencia de asociación(112-117) (Tabla 3). Los resultados fueron dispares también cuando evaluaron la edad de presentación más temprana en repeticiones más cortas CAG(118-120) (Tabla 3).
TABLA 3. Tomado y modificado de Singh R(121). Evaluación comparativa de repeticiones CAG y CP. ND: no disponible.
A diferencia de los estadounidenses, no hay muchos datos para las poblaciones asiáticas y europeas. De los estudios realizados en asiáticos no mostraron asociación con el grado o la edad de diagnóstico en hombres Taiwaneses(121), pero si se asoció a repeticiones más cortas de CAG en hombres japonoses(122), chinos e indios con CP(123-126). Por lo que se puede concluir que hubo correlación entre las secuencias cortas CAG y CP en hombres asiáticos. La mayoría de los estudios en poblaciones europeas no evidenciaron asociación entre la longitud del segmento CAG y riesgo de cáncer de próstata(127-129). Una interpretación de lo dicho hasta aquí podría ser que un número corto de repeticiones de CAG aumenta el riesgo de cáncer de próstata y los resultados disímiles de los estudios se deberían a las diferencias étnicas en la incidencia de cáncer de próstata que a su vez están correlacionados inversamente al número de repeticiones CAG del RA en cada uno de los grupos. Así, los europeos que tienen bajo riesgo de CP presentan largas repeticiones CAG, mientras que en los asiáticos y los de raza negra estadounidenses que tienen una mayor incidencia de CP presentan un bajo número de repeticiones CAG.
D) Mutaciones del RA y cáncer de próstata (CP)
Más allá de los polimorfismos se han descripto mutaciones del RA que se asociaron al mayor riesgo de CP o infertilidad. Ellas involucran principalmente dos regiones del RA: en primer lugar, el dominio de transactivación localizado en el exón 1 donde se encuentran las ya nombradas secuencias polimórficas CAG y GGN y en segundo lugar sustituciones de aminoácidos en el dominio de unión al ligando(130).
Como ya se mencionó, los andrógenos son necesarios para el desarrollo y normal funcionamiento de la próstata. Por lo tanto los moduladores de la actividad del RA siguen siendo importantes blancos en el estudio del CP(131). Aproximadamente el 80-90 % de los CP son dependientes de los andrógenos al inicio del diagnóstico y la endocrinoterapia de dicho cáncer está dirigida a la reducción de andrógenos en el suero y la inhibición de la actividad del RA(132), ya que existen varios estudios que comprobaron que el CP prolifera cuando es expuesto a los Ao y que el RA está involucrado activamente en su progresión(133). Sin embargo, la ablación androgénica puede fallar en última instancia llevando a la progresión del cáncer a un estado refractario a dicha terapéutica, aunque el RA se expresa en toda progresión del CP y persiste en la mayoría de los pacientes con enfermedad resistente al tratamiento hormonal(133,134).
Newmark y col.(135) informó por primera vez una mutación del RA en un paciente con CP. A partir de entonces se han identificado un importante número de mutaciones del gen del RA en dicho cáncer. La mayoría de las mutaciones son sustituciones localizadas en el LBD del RA(133). Hay relativamente pocos informes de los tumores que contienen múltiples mutaciones del RA. El efecto de muchas de las mutaciones halladas todavía no se han investigado in vitro(133). Las consecuencias funcionales más frecuentes de mutaciones aisladas del RA en el CP metastásico es adquirir la capacidad de los antiandrógenos y andrógenos suprarrenales para actuar como agonistas del RA. La mutación T877A del RA permite a los antiandrógenos (hidroxiflutamida y acetato de ciproterona)(136), a la DHEA(137), al androstenediol(133), al estradiol y la progesterona(133,139), a activar la transcripción mediada por el RA. Las mutaciones del RA que le confieren un aumento de la sensibilidad a los andrógenos suprarrenales han sido identificados hasta en el 30 % del CP metastásico estudiados(133).
Así las mutaciones asociadas con el CP llevan a una hiperactividad del receptor mediante la capacidad del RA mutado para responder ‘promiscuamente' a ligandos distintos de los andrógenos testiculares clásicos. La mutación R726L en línea germinal en el LBD no alteró la especificidad de unión al ligando, pero fue activado por estradiol en una medida significativamente mayor que la del receptor de tipo salvaje (sin mutación)(140). El RA mutado en el carcinoma de próstata primario (localizado) también ha mejorado la actividad de transactivación de la androsterona y androstanediol, dos derivados metabólicos de la testosterona presentes en la próstata, así como a la hidroxiflutamida, un antiandrógeno de uso frecuente en la terapia de ablación hormonal. El aumento de la transactivación del receptor mutante no resulta de una alteración en la afinidad del receptor a los ligandos que en condiciones normales lo inducen, ni tampoco de cambios objetivables en la conformación de los receptores que unen el ligando, lo que indica que proteínas correguladoras pueden estar involucradas. Estas mutaciones también eran habituales en el cáncer de próstata metastásico(141). Las mutaciones puntuales en el gen del RA se han identificado en las células metastásicas en cinco de 10 pacientes con cáncer de próstata metastásico. Estudios funcionales de dos de los mutantes del RA demostraron que podría ser activado por la progesterona y estrógeno, proporcionando de este modo una ventaja de crecimiento selectivo de estas células(140).
Por lo tanto la función del RA puede ser alterada por mutaciones en el LBD generando dos patrones funcionales opuestos. Por un lado, una mutación que lleva a una reducción de la transactivación del RA por ligandos normales causante de infertilidad masculina (SIMA) y por el otro, existen mutaciones donde el receptor se activa inapropiadamente por ligandos diferentes a los andrógenos que son las mutaciones encontradas en el CP(142). Es interesante la observación que se realizó en un estudio donde las mutaciones tendrían una localización característica, que explicaría este aumento de transactivación del receptor. De los 27 casos de CP asociado con sustituciones de aminoácidos en el LBD, 74 % (20 casos) se encontraron dentro del LBD agrupado en dos áreas: la primera en la región antes de la 1ra hélice y la segunda entre las hélices 3ra. y 4ta. Estas dos regiones mantienen una proximidad y una relación estrecha y podría formar el sitio de interacciones con proteínas corregulatorias. Comprobaron que mutaciones asociadas con SIA parcial o mínimo y el cáncer de próstata eran mutuamente excluyentes. Esto refuerza la idea de que estas dos condiciones son diferentes extremos de un espectro de mutaciones de una misma región del gen de RA(143).
E) Polimorfismo del RA e infertilidad masculina
Como se viene explicando, el número de repeticiones CAG tendría implicancias en la actividad del RA, por lo tanto es lógico pensar que dada la acción esencial de la To tanto para una adecuada fertilidad en general como para un buen recuento espermático, que una alteración en su acción pueda llevar a una infertilidad. Si bien hay estudios que también mostraron asociación entre criptorquidia (uni o bilateral) y número de repeticiones CAG o GGN, y combinación de ellos(16), no serán motivo de estudio en esta revisión.
Las células germinales son alimentadas y mantenidas por las células de Sertoli para su diferenciación en espermatozoides, a su vez las células de Sertoli dependen de los andrógenos y otras sustancias producidas por las células de Leydig para su adecuado funcionamiento. Esta comunicación paracrina intercelular explica porque a pesar de que las células germinales no expresan el RA, los Ao son esenciales para la correcta espermatogénesis, ya que estos ejercen su acción indirectamente regulando la acción de la célula de Sertoli. Varios estudios mostraron la importancia esencial de los andrógenos en adecuadas concentraciones y actividad, para la diferenciación final posmeiótica de las células germinales hasta llegar a espermátide(139). La To parece jugar un papel en las etapas finales de la diferenciación de los espermatozoides para alcanzar la morfología alargada y por lo tanto las mutaciones puntuales en el gen del RA pueden dar lugar a bajo recuento de espermatozoides, dismórficos (teratozoospermia u oligoteratozoospermia) o directamente a la ausencia de espermatozoides (azoospermia)(144,145).
En un estudio sobre 153 pacientes con alteración en el espermograma, se compararon con 72 controles normales de probada fertilidad(146), mostrando que los pacientes con alteraciones espermáticas presentaba alelos con más de 26 repeticiones CAG, difiriendo significativamente de los controles normales. Los pacientes con 28 o más repeticiones CAG en su gen RA tenían un riesgo cuatro veces mayor de reducción de la espermatogénesis. También hubo una tendencia a presentar un defecto más grave en la espermatogénesis en aquellos con mayor número de repeticiones CAG. Iguales resultados han sido hallados en una población australiana en la que se tomaron 30 pacientes infértiles con azoospermia idiopática u oligozoospermia quienes presentaron repeticiones CAG significativamente más largas que los controles normales(147). Las probabilidades de tener longitudes de 20 repeticiones de CAG fueron seis veces más alto para hombres fértiles que para los hombres con un trastorno espermático. Los datos indican una relación inversa entre la longitud del tracto de poliglutamina del RA y el riesgo de la reducción de la espermatogénesis. Curiosamente, receptores con longitudes de 22 o menos repeticiones de CAG presentaban un mayor riesgo de desarrollar CP(110), pero un riesgo reducido de infertilidad masculina(146). Esto sugiere que la mayor actividad androgénica se asocia a la presencia de alelos del RA con número más corto de repeticiones CAG, que facilita la replicación y adecuada maduración de las células germinales, pero con el riesgo a largo plazo de la sobreestimulación androgénica y consecuente crecimiento de tejido prostático.
Las repeticiones CAG han sido estudiadas en la infertilidad masculina en diversas poblaciones, pero los resultados varían mucho entre ellas. Un aumento de la longitud de la repetición CAG fue asociado con la infertilidad masculina en poblaciones japonesas, americanas, australianas, inglesas, Singapur, francesas y chinas; pero no se halló tal asociación en poblaciones alemanas, belgas, danesas y holandesas(148). La mayoría de los estudios sobre poblaciones europeas no demostraron diferencias significativas en número de repeticiones de CAG entre casos y controles (Tabla 4), excepto dos estudios que informan mayor número de repeticiones en hombres infértiles(149,150) Sin embargo, el análisis combinado de todos los estudios tomados, no mostró diferencias significativas entre hombres europeos fértiles e infértiles(151)(Tabla 4).
TABLA 4. Modificado de Singh R.(121) Evaluación comparativa de repeticiones CAG e infertilidad masculina. ND: no disponible.

F) Mutaciones e infertilidad
Al igual que en cáncer de próstata, más allá de los polimorfismos del RA se describieron mutaciones relacionadas con infertilidad masculina. Involucran principalmente dos regiones del RA: el dominio de transactivación localizado en el exón 1 (donde se encuentran las secuencias polimórficas CAG y GGN) y sustituciones de aminoácidos en el dominio de unión al ligando; dando origen a una insensibilidad mínima a los andrógenos (SIMA), donde su única manifestación es una alteración en la espermatogénesis(130).
Las mutaciones del RA en el LBD que pueden conducir a un SIMA, son las más interesantes, ya que hay posibilidades de corrección hormonal. Se encontró mediante el análisis sistemático del LBD del gen del RA, un polimorfismo conformacional de una sola hebra (SSCP) en más de 200 pacientes con infertilidad masculina idiopática y problemas en la espermatogénesis(152,153). Se identificaron 5 mutaciones exónicas no relacionadas, tres que involucran al codón 886, una en el codón 727 y otra en el 798, todas en el LBD. La mutación M886V se asoció con severa oligospermia y no se la halló en los 400 alelos del RA tomados como controles(154). A pesar que la mutación se encuentra en el LBD del RA, el receptor mutante no altera la unión al andrógeno. Sin embargo, el receptor mutado tiene una capacidad reducida para transactivar los genes blancos andrógeno inducibles. Estos datos sugieren un nuevo mecanismo patogénico por el cual las interacciones defectuosas del receptor con proteínas correguladoras causan enfermedad.
La mutación N727K se produjo en un paciente sin otra evidencia de insensibilidad a los andrógenos pero con un volumen testicular reducido(155). Presentaba oligozoospermia severa con un número de espermatozoides que van entre 2-3 millones por ml. En este caso el paciente fue tratado empíricamente con un análogo de andrógenos, mesterolona, y a los 6 meses de tratamiento el recuento espermático subió a un nivel normal de 28 millones por ml(155). Pudo concebir durante ese período con un bebé sano de término. La mesterolona fue suspendida después de alcanzar la concepción y el número de espermatozoides se redujo a 5 millones por ml. Todo el proceso normal de la espermatogénesis necesita unos 74 días, por lo tanto el tiempo necesario para evaluar un ascenso o diminución del conteo espermático es mayor a los 3 meses de iniciado o suspendido el tratamiento con mesterolona. Estos autores concluyeron que el análogo de andrógenos fue capaz de restablecer la producción normal de espermatozoides, superando su resistencia a mayor concentración. Esto indica que el tratamiento hormonal con análogos de To podría ser útil en la infertilidad masculina cuando es causada por una insensibilidad mínima a los andrógenos(155).
Aunque se han reportado cientos de mutaciones en el gen del RA en diversos trastornos, solo unos pocos se han informado como causa de infertilidad masculina(156). La mayoría de las mutaciones en hombres infértiles dan como resultado la reducción del potencial de transactivación de la proteína mutante. Sin embargo, no se encontró correlación entre el tipo de mutación y el subtipo de la infertilidad (azoospermia, oligozoospermia o oligoteratozoospermia). Se observó la sustitución Gln58Leu en un paciente con azoospermia y en otro con oligoteratozoospermia(157). La especificidad de estas mutaciones y su papel como causa de infertilidad masculina no son claros debido a que en muchos casos se ha informado la existencia de estas mutaciones en pacientes sanos fértiles. Así por ejemplo la mutación Glu211Glu se halló tanto en hombres infértiles como fértiles(158). La reducción del 20 % en el potencial de transactivación del RA, como resultado de la sustitución Gly214Arg resultó en oligozoospermias severas, pero la mutación también se observó en un hombre fértil(159).
Existen estudios que informan el efecto de la mutación sobre la actividad del RA en ensayos in vitro, asociada con la infertilidad o insensibilidad a los andrógenos(156). Así el fenotipo se puede correlacionar con las observaciones in vitro. La sustitución Asn756Ser causó la reducción en un 62 % del potencial de transactivación del RA y resulta en una oligozoospermia severa. Del mismo modo la sustitución Met886Val causa una reducción del 50 % en la transactivación del RA, resultando en una oligozoospermia(160). Un estudio reciente evaluó la secuencia completa de la región codificante del gen del RA en un total 399 hombres infértiles, incluyendo 277 azoospérmicos, 100 oligozoospérmicos y 22 individuos oligoteratozoospérmicos(145). El estudio no reveló mutación alguna del gen del RA en ninguno de los individuos infértiles, por lo que los autores concluyen que las mutaciones del gen de RA podrían contribuir a un porcentaje muy bajo de la infertilidad(145).
G) Fenotipos del Síndrome de Klinefelter y polimorfismos del RA
El fenotipo de presentación del síndrome de Klinefelter es altamente variable, generalmente incluye insuficiencia testicular, deficiencia de andrógenos, pene pequeño, bajo conteo de espermatozoides, talla alta, trastornos cognitivos como alteraciones en el lenguaje, problemas de aprendizaje y dificultad para la lectura(161,162). En un esfuerzo por estudiar los factores que influyen en la variabilidad fenotípica de presentación de los pacientes con síndrome de Klinefelter, Zinn y col. estudiaron el cariotipo para detectar mosaicismo, el genotipo en busca de marcadores microsatélites para determinar el origen de los padres del cromosoma-X supernumerario, el número de repeticiones CAG y velocidad de metilación e inactivación del cromosoma X, en 35 hombres y niños blancos y negros con síndrome de Klinefelter. El estudio mostró que la repetición CAG fue el único factor que influye en el fenotipo del síndrome. La longitud de repeticiones CAG y longitud del pene se correlacionaron inversamente en los individuos afectados(161). Este hallazgo no es sorprendente dada la relación inversa entre longitud de repeticiones CAG y la función de transactivación del RA. Sin embargo se necesitarán más estudios similares para fortalecer tal asociación planteada.
H) Polimorfismo del RA y Síndrome de ovario poliquístico (SOP)
El síndrome de ovario poliquístico (SOP) es un trastorno endocrino caracterizado por la producción y /o actividad anormal de los Ao, que conduce a cambios en el control del desarrollo y maduración folicular(158). Evidencias en primates(164) y mujeres transexuales(165) tratados con altas dosis de andrógenos indican que la morfología característica del ovario poliquístico (numerosos pequeños quistes foliculares en ausencia de folículo dominante) es resultado de la acción directa de los Ao a través de su receptor. Así en las mujeres con SOP, el medio hiperandrogénico del ovario conduce a la anovulación crónica e infertilidad.
Acorde a los datos recabados in vivo, el polimorfismo CAG del receptor androgénico lleva a un aumento de función del receptor que formaría parte de la patogénesis de SOP. Sin embargo los estudios realizados hasta la fecha han mostrado resultados contradictorios(166-169,170). Algunos autores han observado que alelos con un corto número de repeticiones CAG en el RA es más frecuente encontrarlos en mujeres con SOP que en los controles sanos(167). Esto se evidenció también en un estudio donde todas las mujeres SOP presentaron 15 o menos repeticiones CAG(171). Azziz y col. y Schuring y col. reportaron repeticiones significativamente más cortas en pacientes SOP comparada con los controles sanos(166,168). Otro de los estudios que analizaron los dos alelos del RA, refieren una disminución de repeticiones CAG en ambos alelos en mujeres SOP comparado con los controles(172). Mohlig y col. demostraron que en las mujeres con SOP el polimorfismo CAG y su número bajo de repeticiones, tendrían un impacto metabólico como consecuencia del aumento de acción de la To, llevando a la insulinorresistencia, datos que son consistentes con los hallados en estudios funcionales del RA. En evaluaciones estadísticas se informó una variación del 42,5 % del HOMAIR, siendo la To la causa de aumento de la IR en pacientes portadoras de alelos de corta longitud, este efecto se ve atenuado incluso con resultados opuestos en aquellas pacientes con SOP que presentan sus dos alelos con longitudes de 23 o más(173).
Otros estudios no mostraron diferencias entre las repeticiones CAG en pacientes SOP y controles sanos independientemente de otros factores, aunque tomando en consideración ambos alelos juntos(169,174-176). Sin embargo, cuando se compararon los niveles de Ao en el grupo SOP, uno de los estudios mostró una tendencia de repeticiones bialélicas CAG más bajas en el grupo SOP con bajos niveles de To(169). Ello confirma una vez más el rol de la hiperactividad androgénica en el desarrollo del síndrome, sea por hiperandrogenemia o con andrógenos normales pero con aumento de actividad del receptor.
Contrario a la relación inversa entre repeticiones CAG y actividad del RA, un estudio publicado por Hickey mostró que mujeres SOP infértiles presentaban mayor número de repeticiones CAG, con frecuencia eran portadoras de repeticiones CAG bialélicas mayores a 22, comparada con los controles fértiles y la población general(167).
Con respecto al hiperandrogenismo clínico en SOP, los resultados también fueron contradictorios. Varios estudios no encontraron asociación entre el número de repeticiones CAG ni con el hirsutismo (por escala de Ferriman Gallwey)(168,169,176) ni tampoco acné(176). Otros estudios en cambio asociaron el corto número de repeticiones CAG con hirsutismo y acné(170,178,179).
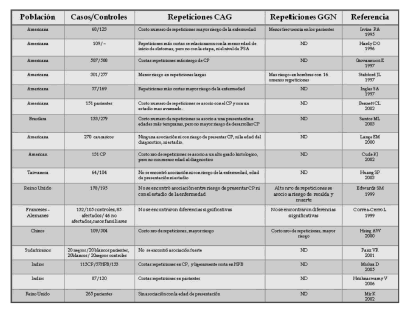
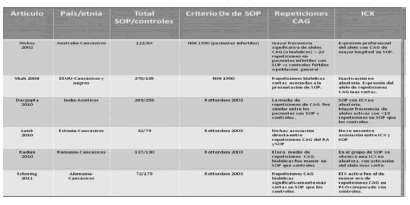
Junto a los datos controvertidos entre la asociación de repeticiones CAG y SOP, debemos considerar la inactivación del X (ICX) como un fenómeno significativo presente en las mujeres, dado que el RA se encuentra en el cromosoma X, por lo tanto la actividad del RA no solo es determinada por el genotipo sino también por efectos epigenéticos de la ICX(170). La metilación del ADN es un importante mecanismo epigenético de inactivación del cromosoma X(180), normalmente ocurre en la vida fetal temprana como un evento azaroso. Una inactivación no azarosa puede ocurrir en cualquiera de los alelos por una exposición ambiental lo que contribuiría a la expresión de SOP(168). Experimentos en modelos animales, en monos y ovejas, mostraron que la exposición al hiperandrogenismo in utero podría ser la causa de anormalidades epigenéticas y reprogramación reproductiva y metabólica tisular del feto produciendo un impacto en el desarrollo neuroendocrino, diferenciación gonadal y organogénesis pancreática(181), resultando en un fenotipo de SOP en la vida adulta(181,182). Los monos expuestos prenatalmente a un ambiente hiperandrogénico, presentaron en la vida adulta un fenotipo símil SOP, con incremento de la adiposidad visceral e insulina elevada(183). La adiposidad visceral tanto en niños como en adultos se correlacionó con los patrones de metilación del ADN(184). Un equivalente natural de fetos expuestos a un ambiente hiperandrogénico in útero, son los hijos de madres SOP. Aunque faltan estudios que determinen si dichos cambios se deberían más bien a los cambios metabólicos que el hiperandrogenismo genera en la madre (ej. insulinorresistencia) que lleva a cambios en el microambiente intrauterino, más que un hiperandrogenismo directo, ya que la alta concentración de aromatasa placentaria no permitiría su paso como tal(181,185-187). Se sabe que el ambiente transitorio hiperandrogénico fetal produce modificaciones en el ADN del feto (tanto en humanos como en monos) produciendo patrones de metilación en varios tejidos, incluyendo la inactivación del cromosoma X llevando a la expresión característica de un solo cromosoma X, generando así un mecanismo crítico para la regulación de la actividad del RA(185-187). Esta hipótesis, donde el ambiente hiperandrogénico puede modificar procesos de metilación, que a su vez llevan a la modificación de la expresión del alelo del RA solo del X activo incluso cuando aparece en la vida adulta, se basaron en estudios que demostraron la pérdida de la metilación del receptor de LH en el ovario en modelos animales con SOP inducido por la administración de DHEA(188). Sin embargo, las controversias también persisten en estudios que analizaron la ICX en pacientes con SOP. (ver tabla 5)
TABLA 5. Evaluación comparativa de repeticiones CAG y SOP. Tomado de Baculescu N.(38)
Todo lo dicho hasta aquí nos permite concluir que existen 3 grandes limitaciones de los estudios de asociación del polimorfismo CAG y SOP:
1. Número de repeticiones CAG étnicos y criterios diagnósticos de SOP: a) por un lado los resultados inconsistentes se pueden explicar, al menos en parte, por los diferentes orígenes étnicos de las personas tomadas en cada estudio, sean del grupo con SOP o como controles(165,169,177). El rango normal de repeticiones de CAG varía de acuerdo a la etnia, aproximadamente 10 a 30 en los caucásicos(184), un poco más corto en los afroamericanos y más largo en los asiáticos(190,191). Más importante aún si los sujetos control fueron reclutados en una población diferente en comparación con la grupos correspondientes con SOP(169,171,174). b) Otros de los factores de confusión pueden ser los diferentes criterios utilizado para definir SOP (NIH 1990 o Rotterdam 2003), como también el tamaño muestral, sea de pacientes o de controles.
2. Cambios epigenéticos: a) En la actualidad solo dos sitios de metilación han sido identificados y evaluados en el análisis del gen del RA por técnica de PCR, después de la digestión de ADN con methylation-sensitive Hpall(166,168,172,177,179). Los resultados obtenidos informan que la ICX no fue significativamente diferente entre pacientes con síndrome de ovario poliquístico y controles. Sin embargo, tres importantes estudios han encontrado que el alelo más corto del RA era el preferentemente activo en el subgrupo con SOP mostrando una ICX no aleatoria(166,168,179). Otros estudios mostraron que la combinación bialélica presenta un número de repeticiones más baja, e incluso la expresión del X no sería aleatoria en paciente con SOP. En el SOP hirsuto, un estudio concluyó que el hirsutismo normoandrogénico se asoció con metilación preferencial del alelo más largo de los dos del RA, lo que permite la expresión solo del más corto (y presumiblemente el alelo más funcional) que se expresa en el cromosoma X activo(192). Sin embargo, no hay diferencia en el patrón de inactivación del X en otros estudios de SOP hirsutas y sin hirsutismo(166,168,193). Lo que sí observaron en uno de ellos, fue una discreta preferencia de expresión del alelo CAG más corto(168). En este contexto, la información suplementaria puede ser obtenida mediante la investigación de los patrones de metilación de los sitios completos del RA cerca de las repeticiones polimórficas CAG porque todos esos sitios CpG pueden estar involucrados en regulación de la expresión del RA en el cromosoma X activo(188). b) Por otro lado, debemos considerar que la ICX puede variar en los diferentes tejidos. En todos los estudios nombrados anteriormente, el ICX fue generalmente evaluado en linfocitos de sangre periférica, que no parecen reflejar la acción de los andrógenos en tejidos diana(165). Un importante estudio demostró que la metilación del ADN de leucocitos de sangre periférica no se altera en pacientes con SOP vs. los controles(194).
3. Interacción del RA con coactivadores y correpresores, estos han sido identificados solo en parte y se consideran críticos para la función del RA, dado que median el efecto andrógeno específico de tejido. Una variación en la expresión o función de estos correguladores pueden contribuir al desarrollo de SOP(168,195). Es necesario en un futuro realizar estudios genéticos y fisiológicos para identificar las interacciones funcionales entre las repeticiones de CAG y los correguladores del RA(168).
CONCLUSIÓN
Se puede concluir que en patologías andrógenos dependientes es importante la evaluación de la actividad del RA como determinante fisiopatológico de las mismas; pero querer encarar las implicancias fisiopatológicas del RA solo a partir de las mutaciones y polimorfismos del receptor, es por ahora ilusorio, dado que la acción del receptor no solo depende de la interacción hormona-receptor, sino también del medio (tejido) en el que este actúa. Es decir de la presencia de correguladores, de la concentración de los mismos en el tejido, de polimorfismos en el promotor del gen diana de Ao, y otros tantos factores, aún no del todo conocidos, que determinan la actividad del RA. Además existen características polimórficas propias de cada etnia que dificultan la interpretación de los resultados obtenidos en cada estudio, como es la conocida diferencia entre el número de repeticiones CAG del RA en diferentes grupos poblacionales y su asociación con patologías como el CP, infertilidad y SOP. Más complejo es aún si se tienen en cuenta los dos alelos del RA en la mujer y su expresión variable por un mecanismo de ICX. Este sin fin de determinantes de la actividad del RA es lo que probablemente explica en parte, la variabilidad y muchas veces discordancia de los resultados hallados en los diferentes análisis.
Por todo lo dicho, si bien las mutaciones y los polimorfismos determinan un aumento o disminución de la actividad del RA sin modificación en la concentración de andrógenos séricos, solo explican en parte la fisiopatología de las enfermedades andrógeno-dependientes con niveles séricos normales de Ao, siendo probablemente la suma de factores lo que determina su aparición.
1. Haqq C, Donahoe P. Regulation of sexual dimorphism in mammals. Physiol Rev 78:1-33, 1998 [ Links ]
2. Sinha-Hikim I, Taylor W, Gonzalez-Cadavid N, Zheng W, Bhasin S. Androgen receptor in human skeletal muscle and cultured muscle satellite cells:up-regulation by androgen treatment. J Clin Endocrinol Metab 89:5245-55, 2004 [ Links ]
3. Eysenck H, Eysenck S. Psychoticism as a Dimension of Personity. London:Hodder and Stoughton. [ Links ]
4. Giammanco M, Tabacchi G, Giammanco S, Di Majo D, La Guardia M. Testosterone and aggressiveness. Med Sci Monit 11:RA136-45, 2005 [ Links ]
5. Wang C, Alexander G, Berman N, SalehianB, Davidson T, McDonald V, Steiner B, Hull L, Callegari C, Swerdloff RS. Testosterone replacement therapy improves mood in hypogonadal men - a clinical research center study. J Clin Endocrinol Metab 81:3578-83, 1996 [ Links ]
6. Singh R, Artaza J, Taylor W, et al. Testosterone inhibits adipogenic differentiation in 3T3-L1 cells:nuclear translocation of androgen receptor complex with beta-catenin and T-cell factor 4 may bypass canonical Wnt signaling to down-regulate adipogenic transcription factors. Endocrinology 147:141-4, 2006 [ Links ]
7. Shiina H, Matsumoto T, Sato T, Igarashi K, Miyamoto J, Takemasa S, Sakari M, Takada I, Nakamura T, Metzger D, Chambon P, Kanno J, Yoshikawa H, Kato S. Premature ovarian failure in androgen receptor-deficient mice, Proc Natl Acad Sci USA 103:224-29, 2006 [ Links ]
8. Casson P, Toth M, Johnson J, Stanczyk F, Casey C, Dixon M. Correlation of Serum Androgens with Anthropometric and metabolic Indices in Healthy, Nonobese Postmenopausal Women. J Clin Endocrinol Metab 95:4276-82, 2010 [ Links ]
9. Quigley C, De Bellis A, Marschke K, El Awady M, Wilson E, French F. Androgen receptor defects:historical, clinical and molecular perspectives. Endocr Rev 16:271-321, 1995 [ Links ]
10. Chang AR. Structure. Experimental Clinical Endocrinol Diabetes. 1988 [ Links ]
11. Beato M, Herrlich P, Schutz G. Steroid-Hormone Receptors-Many Actors in Search of A Plot. Cell 83:851-57, 1995 [ Links ]
12. Brinkmann A. Molecular basis ofandrogen insensitivity. Mol Cel Endocrinol 179:105-09, 2001 [ Links ]
13. Faber P, Kuiper G, van Rooij H, van der Korput J, Brinkmann A, Trapman J. The N-terminal domain of the human androgen receptor is encoded by one, large exon. Mol Cell Endocrinol 61:257-62, 1989 [ Links ]
14. Chamberlain N, Driver E, Miesfeld R. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res 22:3181-86, 1994 [ Links ]
15. Kazemi-Esfarjani P, Trifiro MA, Pinsky L. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor:possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum Mol Genet 4:523-27, 1995 [ Links ]
16. Lim H, Nixon R, Chen H, Hughes I, Hawkins J. Evidence that longer androgen receptor polyglutamine repeats are a causal factor for genital abnormalities. J Clin Endocrinol Metab 86:3207-10, 2001 [ Links ]
17. Lubahn D, Joseph D, Sullivan P, Willard H, French F, Wilson E. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science 240:327-30, 1988 [ Links ]
18. Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Bäsler S, Schäfer M, Egner U, Carrondo MA. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem 275:26164-71, 2000 [ Links ]
19. Brinkmann A, Faber P, van Rooij H, Kuiper G, Ris C, Klaassen P, et al. The human androgen receptor domain structure, genomic organization and regulation of expression. J Steroid Biochem 34:307-10, 1989 [ Links ]
20. Ivell R, Hartung S. The molecular basis of cryptorchidism. Mol Hum Reprod 9:175-81, 2003 [ Links ]
21. Brinkmann AO. Molecular basis of androgen insensitivity. Mol Cell Endocrinol 179:105-19, 2001 [ Links ]
22. Dehm S, Tindall D. Androgen receptor structural and functional elements:Role and regulation in prostate cancer. Mol Endocrinol 21:2855-63, 2007 [ Links ]
23. Newmark J. Androgen receptor gene mutations in human prostate cancer. Proc Natl Acad Sci 89:6319-23, 1992 [ Links ]
24. Kohler B, Lumbroso S, Leger J, Audran F, Grau ES, Kurtz F, Pinto G, Salerno M, Semitcheva T, Czernichow P, Sultan C. Androgen insensitivity syndrome:Somatic mosaicism of the androgen receptor in seven families and consequences for sex assignment and genetic counseling. J Clin Endocrinol Metab 90:106-11, 2005 [ Links ]
25. Chamberlain N, Driver E, Miesfeld R. The Length and Location of Cag Trinucleotide Repeats in the Androgen Receptor N-Terminal Domain Affect Transactivation Function. Nucleic Acids Res 22:3181-86, 1994 [ Links ]
26. Tut T, Ghadessy F, Trifiro M, Pinsky L, Yong E. Long polyglutamine tracts in the androgen receptor are associated with reduced transactivation, impaired sperm production, and male infertility. J Clin Endocrinol Metab 82:3777-82, 1997 [ Links ]
27. Choong C, Kemppainen J, Zhou Z, Wilson E. Reduced androgen receptor gene expression with first exon CAG repeat expansion. Mol Endocrinol 10(12):1527-35, 1996 [ Links ]
28. Nenonen. CAG repeat number is not inversely associated with androgen receptor activity in vitro. Mol Hum Reprod 16:153-57, 2010 [ Links ]
29. Fischbeck K, Lieberman A, Bailey C, Abel A, Merry D. Androgen receptor mutation in Kennedy's disease. Philosophical Transactions of the Royal Society of London Series B-Biological Sciences. 354(1386):1075-78, 1999 [ Links ]
30. Zitzmann M, Depenbusch M, Gromoll J, Nieschlag E. Prostate volume and growth in testosterone-substituted hypogonadal men are dependent on the CAG repeat polymorphism of the androgen receptor gene:A longitudinal pharmacogenetic study. J Clin Endocrinol Metab 88:2049-54, 2003 [ Links ]
31. Zitzmann M, Depenbusch M, Gromoll J, Nieschlag E. X-chromosome inactivation patterns and androgen receptor functionality influence phenotype and social characteristics as well as pharmacogenetics of testosterone therapy in Klinefelter patients. J Clin Endocrinol Metab 89:6208-17, 2004 [ Links ]
32. Zinn A, Ramos P, Elder F, Kowal K, Samango-Spouse C, Ross J. Androgen receptor CAG(n) repeat length influences phenotype of 47,XXY (Klinefelter) syndrome. J Clin Endocrinol Metabolism 90:5041-46, 2005 [ Links ]
33. Hughes I, Houk C, Ahmed S, Lee P, and the LWPES Consensus Group, and the ESPE Consensus Group. Consensus statement on management of intersex disorders. Arch Dis Child 91:554-63, 2006 [ Links ]
34. Pasterski V, Prentice P, Hughes I. Consequences of the Chicago consensus on disorders of sex development (DSD):current practices in Europe. Arch Dis Child 95:618-23, 2010 [ Links ]
35. Hughes I. The quiet revolution:disorders of sex development. Best Pract Res Clin Endocrinol Metab 24:159-62, 2010 [ Links ]
36. Achermann J, Hughes I. Disorders of sex development. In:Kronenberg H, Melmed S, Polonsky K, Larsen P, eds. Williams Textbook of Endocrinology, 12th ed. Philadelphia:Saunders Elsevier, 863-94, 2011 [ Links ]
37. Ieuan A Hughes, John D Davies, Trevor I Bunch, Vickie Pasterski, Kiki Mastroyannopoulou, Jane MacDougall. Androgen insensitivity syndrome. Seminar Lancet 380:1419-28, 2012 [ Links ]
38. Baculescu N. The role of androgen receptor activity mediated by the CAG repeat polymorphism in the pathogenesis of PCOS. J Med Life 6:18-25, 2013 [ Links ]
39. Morris J. The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol 65:1192-1211, 1953 [ Links ]
40. Quigley C, De Bellis A, Marschke K, el-Awady M, Wilson E, French F. Androgen receptor defects:historical, clinical, and molecular perspectives. Endocr Rev 16:271-321, 1995 [ Links ]
41. Boehmer A, Brinkmann O, Brüggenwirth H, et al. Genotype versus phenotype in families with androgen insensitivity syndrome. J Clin Endocrinol Metab 86:4151-60, 2001 [ Links ]
42. Sarpel U, Palmer S, Dolgin S. The incidence of complete androgen insensitivity in girls with inguinal hernias and assessment of screening by vaginal length measurement. J Pediatr Surg 40:133-37, 2005 [ Links ]
43. Hurme T, Lahdes-Vasama T, Mäkela E, Iber T, Toppari J. Clinical findings in prepubertal girls with inguinal hernia with special reference to the diagnosis of androgen insensitivity syndrome. Scand J Urol Nephrol 43:42-46, 2009 [ Links ]
44. Deeb A, Hughes I. Inguinal hernia in female infants:a cue to check the sex chromosomes? BJU Int 96:401-03, 2005 [ Links ]
45. Chiu R, Lo Y, Lo D. Non-invasive prenatal diagnosis by fetal nucleic acid analysis in maternal plasma:the coming of age. Semin Fetal Neonatal Med 16:88-93, 2011 [ Links ]
46. Yalinkaya A, Yayla M, Erdemoglu M. Prenatal diagnosis of a fetus with androgen insensitivity syndrome (AIS). Prenat Diagn 27:856-57, 2007 [ Links ]
47. Stewart C, Baker E, Beaton C, Crook M, Peverall J, Wallace S. Detection of Y-chromosome in gonadal tumours using fl uorescence in situ hybridization:diagnostic value in intersex conditions including older patients with clinically unsuspected androgen insensitivity syndrome. Histopathology 52:175-82, 2008 [ Links ]
48. Han TS, Goswami D, Trikudanathan S, Creighton SM, Conway GS. Comparison of bone mineral density and body proportions between women with complete androgen insensitivity syndrome and women with gonadal dysgenesis. Eur J Endocrinol 159:179-85, 2008 [ Links ]
49. Papadimitriou DT, Linglart A, Morel Y, Chaussain J-L. Puberty in subjects with complete androgen insensitivity syndrome. Horm Res 65:126-31, 2006 [ Links ]
50. Danilovic D, Correa P, Costa E, Melo K, Mendonca B, Arnhold I. Height and bone mineral density in androgen insensitivity syndrome with mutations in the androgen receptor gene. Osteoporos Int 18:369-74, 2007 [ Links ]
51. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, Ferreira T, Wood AR, Weyant RJ, Segrè AV, Speliotes EK, Wheeler E, Soranzo N, Park JH, Yang J, Gudbjartsson D, Heard-Costa NL, Randall JC, Qi L, Vernon Smith A, Mägi R, Pastinen T, Liang L, Heid IM, Luan J, Thorleifsson G, Winkler TW, Goddard ME, Sin Lo K, Palmer C, Workalemahu T, Aulchenko YS, Johansson A, Zillikens MC, Feitosa MF, Esko T, Johnson T, Ketkar S, Kraft P, Mangino M, Prokopenko I, Absher D, Albrecht E, Ernst F, Glazer NL, Hayward C, Hottenga JJ, Jacobs KB, Knowles JW, Kutalik Z, Monda KL, Polasek O, Preuss M, Rayner NW, Robertson NR, Steinthorsdottir V, Tyrer JP, Voight BF, Wiklund F, Xu J, Zhao JH, Nyholt DR, Pellikka N, Perola M, Perry JR, Surakka I, Tammesoo ML, Altmaier EL, Amin N, Aspelund T, Bhangale T, Boucher G, Chasman DI, Chen C, Coin L, Cooper MN, Dixon AL, Gibson Q, Grundberg E, Hao K, Juhani Junttila M, Kaplan LM, Kettunen J, König IR, Kwan T, Lawrence RW, Levinson DF, Lorentzon M, McKnight B, Morris AP, Müller M, Suh Ngwa J, Purcell S, Rafelt S, Salem RM, Salvi E, Sanna S, Shi J, Sovio U, Thompson JR, Turchin MC, Vandenput L, Verlaan DJ, Vitart V, White CC, Ziegler A, Almgren P, Balmforth AJ, Campbell H, Citterio L, De Grandi A, Dominiczak A, Duan J, Elliott P, Elosua R, Eriksson JG, Freimer NB, Geus EJ, Glorioso N, Haiqing S, Hartikainen AL, Havulinna AS, Hicks AA, Hui J, Igl W, Illig T, Jula A, Kajantie E, Kilpeläinen TO, Koiranen M, Kolcic I, Koskinen S, Kovacs P, Laitinen J, Liu J, Lokki ML, Marusic A, Maschio A, Meitinger T, Mulas A, Paré G, Parker AN, Peden JF, Petersmann A, Pichler I, Pietiläinen KH, Pouta A, Ridderstråle M, Rotter JI, Sambrook JG, Sanders AR, Schmidt CO, Sinisalo J, Smit JH, Stringham HM, Bragi Walters G, Widen E, Wild SH, Willemsen G, Zagato L, Zgaga L, Zitting P, Alavere H, Farrall M, McArdle WL, Nelis M, Peters MJ, Ripatti S, van Meurs JB, Aben KK, Ardlie KG, Beckmann JS, Beilby JP, Bergman RN, Bergmann S, Collins FS, Cusi D, den Heijer M, Eiriksdottir G, Gejman PV, Hall AS, Hamsten A, Huikuri HV, Iribarren C, Kähönen M, Kaprio J, Kathiresan S, Kiemeney L, Kocher T, Launer LJ, Lehtimäki T, Melander O, Mosley TH Jr, Musk AW, Nieminen MS, O'Donnell CJ, Ohlsson C, Oostra B, Palmer LJ, Raitakari O, Ridker PM, Rioux JD, Rissanen A, Rivolta C, Schunkert H, Shuldiner AR, Siscovick DS, Stumvoll M, Tönjes A, Tuomilehto J, van Ommen GJ, Viikari J, Heath AC, Martin NG, Montgomery GW, Province MA, Kayser M, Arnold AM, Atwood LD, Boerwinkle E, Chanock SJ, Deloukas P, Gieger C, Grönberg H, Hall P, Hattersley AT, Hengstenberg C, Hoffman W, Lathrop GM, Salomaa V, Schreiber S, Uda M, Waterworth D, Wright AF, Assimes TL, Barroso I, Hofman A, Mohlke KL, Boomsma DI, Caulfield MJ, Cupples LA, Erdmann J, Fox CS, Gudnason V, Gyllensten U, Harris TB, Hayes RB, Jarvelin MR, Mooser V, Munroe PB, Ouwehand WH, Penninx BW, Pramstaller PP, Quertermous T, Rudan I, Samani NJ, Spector TD, Völzke H, Watkins H, Wilson JF, Groop LC, Haritunians T, Hu FB, Kaplan RC, Metspalu A, North KE, Schlessinger D, Wareham NJ, Hunter DJ, O'Connell JR, Strachan DP, Wichmann HE, Borecki IB, van Duijn CM, Schadt EE, Thorsteinsdottir U, Peltonen L, Uitterlinden AG, Visscher PM, Chatterjee N, Loos RJ, Boehnke M, McCarthy MI, Ingelsson E, Lindgren CM, Abecasis GR, Stefansson K, Frayling TM, Hirschhorn JN. Hundreds of variants clustered in genomic loci and biological pathways aff ect human height. Nature 467: 832-38, 2010 [ Links ]
52. Miles H, Gidlöf S, Nordenström A, Ong K, Hughes I. The role of androgens in fetal growth:observational study in two genetic models of disordered androgen signalling. Arch Dis Child Fetal Neonatal Ed 95:F435-38, 2010 [ Links ]
53. Ahmed S, Achermann J, Arlt W, Balen A, Conway G, Edwards Z, Elford S, Hughes I, Izatt L, Krone N, Miles H, O'Toole S, Perry L, Sanders C, Simmonds M, Wallace A, Watt A, Willis D. UK guidance on the initial evaluation of an infant of an adolescent with a suspected disorder of sex development. Clin Endocrinol 75(1):12-26, 2011 [ Links ]
54. Veiga-Junior N, Medeats P, Petroli R, Calais F, de Mello M, Castro C, Guaragna-Filho G, Sewaybricker L, Marques-de-Faria A, Maciel-Guerra A, Guerra-Junior G. Clinical and laboratorial features that may differentiate 46,XY DSD due to partial androgen insensitivity and 5a-reductase type 2 deficiency. Int J Endocrinol, published online Dec 12. DOI:10.1155/2012/964876, 2012 [ Links ]
55. Zuccarello D, Ferlin A, Vinanzi C, Prana E, Garolla A, Callewaert L, Claessens F, Brinkmann AO, Foresta C. Detailed functional studies on androgen receptor mild mutations demonstrate their association with male infertility. Clin Endocrinol (Oxf) 68:580-88, 2008 [ Links ]
56. Yong E, Ng S, Roy A, Yun G, Ratnam S. Pregnancy after hormonal correction of severe spermatogenic defect due to mutation in androgen receptor gene. Lancet 344:826-27, 1994 [ Links ]
57. Wang R, Yeh S, Tzeng C, Chang C. Androgen receptor roles in spermatogenesis and fertility:lessons from testicular cell-specific androgen receptor knockout mice. Endocr Rev 30:119-32, 2009 [ Links ]
58. Finsterer J. Bulbar and spinal muscular atrophy (Kennedy'sdisease):a review. Eur J Neurol 16:556-61, 2009 [ Links ]
59. Melo K, Mendonca B, Billerbeck A, Costa E, Inácio M, Silva F, Leal A, Latronico A, Arnhold I. Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort:fi ve novel mutations in the androgen receptor gene. J Clin Endocrinol Metab 88:3241-50, 2003 [ Links ]
60. Hughes I, Deeb A. Androgen resistance. Best Pract Res Clin Endocrinol Metab 20:577-98, 2006 [ Links ]
61. Sinnecker G, Hiort O, Nitsche E, Holterhus P, Kruse K, and the German Collaborative Intersex Study Group. Functional assessment and clinical classifi cation of androgen sensitivity in patients with mutations of the androgen receptor gene. Eur J Pediatr 156:7-14, 1997 [ Links ]
62. Bouvattier C, Carel J, Lecointre C, David A, Sultan C, Bertrand A, Morel Y, Chaussain J. Postnatal changes of T, LH, and FSH in 46,XY infants with mutations in the AR gene. J Clin Endocrinol Metab 87:29-32, 2002 [ Links ]
63. Quigley C. Editorial:The postnatal gonadotropin and sex steroid surge-insights from the androgen insensitivity syndrome. J Clin Endocrinol Metab 87:24-28, 2002 [ Links ]
64. Bergadá I, Milani C, Bedecarrás P, Andreone L, Ropelato MG, Gottlieb S, Bergadá C, Campo S, Rey R. Time course of the serum gonadotropin surge, inhibins, and anti-Müllerian hormone in normal newborn males during the fi rst month of life. J Clin Endocrinol Metab 91:4092-98, 2006 [ Links ]
65. Bay K, Virtanen H, Hartung S, Ivell R, Main K, Skakkebaek N, Andersson A; Nordic Cryptorchidism Study Group, Toppari J. Insulin-like factor 3 levels in cord blood and serum from children:eff ects of age, postnatal hypothalamic-pituitary-gonadal axis activation, and cryptorchidism. J Clin Endocrinol Metab 92:4020-27, 2007 [ Links ]
66. Ahmed S, Keir L, McNeilly J, Galloway P, O'Toole S, Wallace A. The concordance between serum anti-Mullerian hormone and testosterone concentrations depends on duration of hCG stimulation in boys undergoing investigation of gonadal function. Clin Endocrinol (Oxf) 72:814-19, 2010 [ Links ]
67. Aksglaede L, Sørensen K, Boas M, Mouritsen A, Hagen C, Jensen R, Petersen J, Linneberg A, Andersson A, Main K, Skakkebæk N, Juul A. Changes in anti-Müllerian hormone (AMH) throughout the life span:a population-based study of 1027 healthy males from birth (cord blood) to the age of 69 years. J Clin Endocrinol Metab 95:5357-64, 2010 [ Links ]
68. Rey R, Mebarki F, Forest M, Mowszowicz I, Cate R, Morel Y, Chaussain J, Josso N. Anti-müllerian hormone in children with androgen insensitivity. J Clin Endocrinol Metab 79:960-64, 1994 [ Links ]
69. Boukari K, Meduri G, Brailly-Tabard S, Guibourdenche J, Ciampi M, Massin N, Martinerie L, Picard J, Rey R, Lombès M, Young J.
Lack of androgen receptor expression in Sertoli cells accounts for the absence of anti-mullerian hormone repression during early human testis development. J Clin Endocrinol Metab 94:1818-25, 2009
70. Michala L, Goswami D, Creighton S, Conway G. Swyer syndrome:presentation and outcomes. BJOG 115:737-41, 2008 [ Links ]
71. Pittock S, Babovic-Vuksanovic D, Lteif A. Mayer-Rokitansky-Küster-Hauser anomaly and its associated malformations. Am J Med Genet A 135:314-16, 2005 [ Links ]
72. Grinsponand R, Rey R. “Anti-Mullerian hormone and sertoli cell function in paediatric male hypogonadism,” Horm Rese Paediatr 73:81-92, 2010
73. Gottlieb B, Beitel, L, Lumbroso R, Pinsky L, Trifiro M. Update of the androgen receptor gene mutations database. Hum Mutat 14, 103-114, 1999 [ Links ]
74. Matias P, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Bäsler S, Schäfer M, Egner U, Carrondo M. Structural evidence for ligand specifi city in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem 275:26164-71, 2000 [ Links ]
75. Wang M, Wang J, Zhang Z, Zhao Z, Zhang R, Hu X, Tan T, Luo S, Luo Z. Dissecting phenotypic variation among AIS patients. Biochem Biophys Res Commun 335:335-42, 2005 [ Links ]
76. Holterhus P, Deppe U, Werner R, Richter-Unruh A, Bebermeier J, Wünsch L, Krege S, Schweikert H, Demeter J, Riepe F, Hiort O, Brooks J. Intrinsic androgen-dependent gene expression patterns revealed by comparison of genital fibroblasts from normal males and individuals with complete and partial androgen insensitivity syndrome. BMC Genomics 8:376, 2007 [ Links ]
77. Szafran A, Hartig S, Sun H, Shen Y, Mediwala S, Bell J, McPhaul M, Mancini M, Marcelli M. Androgen receptor mutations associated with androgen insensitivity syndrome:a high content analysis approach leading to personalized medicine. PLoS One 4:e8179, 2009 [ Links ]
78. Lundberg Giwercman Y, Nikoshkov A, Lindsten K, Byström B, Pousette A, Knudtzon J, Alm J, Wedell A. Response to treatment in patients with partial androgen insensitivity due to mutations in the DNA-binding domain of the androgen receptor. Horm Res 53:83-88, 2000 [ Links ]
79. Tadokoro R, Bunch T, Schwabe J, Hughes I, Murphy J. Comparison of the molecular consequences of different mutations at residue 754 and 690 of the androgen receptor (AR) and androgen insensitivity syndrome (AIS) phenotype. Clin Endocrinol (Oxf) 71:253-60, 2009 [ Links ]
80. Leslie N. Haldane was right:de novo mutations in androgen insensitivity syndrome. J Pediatr 132:939-43, 1998 [ Links ]
81. Köhler B, Lumbroso S, Leger J, Audran F, Grau E, Kurtz F, Pinto G, Salerno M, Semitcheva T, Czernichow P, Sultan C. Androgen insensitivity syndrome:somatic mosaicism of the androgen receptor in seven families and consequences for sex assignment and genetic counseling. J Clin Endocrinol Metab 90:106-11, 2005 [ Links ]
82. Evans B, Hughes I, Bevan C, Patterson M, Gregory J. Phenotypic diversity in siblings with partial androgen insensitivity syndrome. Arch Dis Child 76:529-31, 1997 [ Links ]
83. Holterhus P, Sinnecker G, Hiort O. Phenotypic diversity and testosterone-induced normalization of mutant L712F androgen receptor function in a kindred with androgen insensitivity. J Clin Endocrinol Metab 85:3245-50, 2000 [ Links ]
84. Cheikhelard A, Morel Y, Thibaud E, Lortat-Jacob S, Jaubert F, Polak M, Nihoul-Fekete C. Long-term followup andcompar ison between genotype and phenotype in 29 cases of complete androgen insensitivity syndrome. J Urol 180:1496-501, 2008 [ Links ]
85. Yarbrough W, Quarmby V, Simental J, Joseph DR, Sar M, Lubahn D, Olsen K, French F, Wilson E. A single base mutation in the androgen receptor gene causes androgen insensitivity in the testicular feminized rat. J Biol Chem 265:8893-900, 1990 [ Links ]
86. Ahmed S, Cheng A, Dovey L, Hawkins J, Martin H, Rowland J, Shimura N, Tait AD, Hughes I. Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab 85:658-65, 2000 [ Links ]
87. Werner R, Zhan J, Gesing J, Struve D, Hiort O. In-vitro characterization of androgen receptor mutations associated with complete androgen insensitivity syndrome reveals distinct functional deficits. Sex Dev 2:73-83, 2008 [ Links ]
88. Hughes I, Evans B. Complete androgen insensitivity syndrome characterized by increased concentration of a normal androgen receptor in genital skin fi broblasts. J Clin Endocrinol Metab 63:309-15, 1986 [ Links ]
89. Quigley C, Evans B, Simental J, Marschke K, Sar M, Lubahn D, Davies P, Hughes I, Wilson E, French F. Complete androgen insensitivity due to deletion of exon C of the androgen receptor gene highlights the functional importance of the second zinc finger of the androgen receptor in vivo. Mol Endocrinol 6:1103-12, 1992 [ Links ]
90. Wooster R, Mangion J, Eeles R, Smith S, Dowsett M, Averill D, Barrett-Lee P, Easton D, Ponder B, Stratton M. A germ-line mutation in the androgen receptor gene in two brothers with breast cancer and Reifenstein syndrome. Nat Genet 2, 132-34, 1992 [ Links ]
91. Hiort O, Wodtke A, Struve D, Zöllner A, Sinnecker G, and the German Collaborative Intersex Study Group. Detection of point mutations in the androgen receptor gene using non-isotopic single strand conformation polymorphism analysis. Hum Mol Genet 3:1163-66, 1994 [ Links ]
92. Scheiber D, Barta C, Halász Z, Sallai A, Rácz K, Ságodi L, Fekete G, Hiort O, Sólyom J. Mutational analysis of Hungarian patients with androgen insensitivity syndrome. J Pediatr Endocrinol Metab 16:367-73, 2003 [ Links ]
93. Audi L, Fernández-Cancio M, Carrascosa A, Andaluz P, Torán N, Piró C, Vilaró E, Vicens-Calvet E, Gussinyé M, Albisu MA, Yeste D, Clemente M, Hernández de la Calle I, Campo M Del, Vendrell T, Blanco A, Martínez-Mora J, Granada ML, Salinas I, Forn J, Calaf J, Angerri O, Martínez-Sopena M J, Valle J Del, García E, Gracia-Bouthelier R, Lapunzina P, Mayayo E, Labarta JI, Lledó G, Sánchez Del Pozo J, Arroyo J, Pérez-Aytes A, Beneyto M, Segura A, Borrás V, Gabau E, Caimarí M, Rodríguez A, Martínez-Aedo M J, Carrera M, Castaño L, Andrade M, Bermúdez de la Vega J A, and the Grupo de Apoyo al Síndrome de Insensibilidad a los Andrógenos (GrApSIA). Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. J Clin Endocrinol Metab 95:187688, 2010 [ Links ]
94. Zuccarello D, Ferlin A, Vinanzi C, Prana E, Garolla A, Callewaert L, Claessens F, Brinkmann A, Foresta C. Detailed functional studies on androgen receptor mild mutations demonstrate their association with male infertility. Clin Endocrinol (Oxf) 68:580-88, 2008 [ Links ]
95. Arbizu T, Santamaria J, Gomex J, Quilez A, Serra J. A family with adult spinal and bulbar muscular atrophy, Xlinked inheritance and associated testicular failure. J Neurol Sci 59:371-82, 1983 [ Links ]
96. Amato A, Prior T, Barohn R, Snyder P, Papp A, Mendell J. Kennedy's disease:a clinicopathologic correlation with mutations in the androgen receptor gene. Neurology 43:791-94, 1993 [ Links ]
97. Santarosa M, Bidoli E, Gallo A, Steffan A, Boiocchi M, Viel A. Polymorphic CAG repeat length within the androgen receptor gene:identification of a subgroup of patients with increased risk of ovarian cancer. Oncol Rep 9:639-44, 2002 [ Links ]
98. Spurdle A, Webb PM, Chen X, Martin N, Giles G, Hopper J, Chenevix-Trench G. Androgen receptor exon 1 CAG repeat length and risk of ovarian cancer. Int J Cancer 87:637-43, 2000 [ Links ]
99. Hogervorst E, Williams J, Budge M, Barnetson L, Combrinck M, Smith A. Serum total testosterone is lower in men with Alzheimer's disease. Neuroendocrinol Lett 3:163-8, 2001 [ Links ]
100. Turakulov R, Jorm A, Jacomb P, TanX, Easteal S. Association of dopamine-b-hydroxylase and androgen receptor gene polymorphisms with Eysenck's P and other personality traits. Pers Individ Dif 37:191-202, 2004 [ Links ]
101. Gottlieb B, Beitel L, Wu J, Trifiro M. The androgen receptor gene mutations database (ARDB):2004 update. Hum Mutat 23:527-33, 2004 [ Links ]
102. Hsiao P, Lin D, Nakao R. The linkage of Kennedy's neuron disease to ARA24, the first identified androgen receptor polyglutamine region-associated coactivator. J Biol Chem 274:20229-34, 1999 [ Links ]
103. Irvine R, Ma H, Yu M, Ross R, Stallcup M, Coetzee G. Inhibition of p160 mediated coactivation with increasing androgen receptor polyglutamine length. Hum Mol Genet 9:267-74, 2000 [ Links ]
104. Eu Leong Yong, Joyce Lim, Wang Qi, Victor Ong and Amparo Mifsud. Molecular basis of androgen receptor diseases. Ann Med 32:15-22, 2000 [ Links ]
105. Nance M. Clinical aspects of CAG repeat diseases. Brain Pathol 7, 881-900, 1997 [ Links ]
106. Stenoien D, Cummings C, Adams H, Mancini M, Patel K, DeMartino G, Marcelli M, Weigel N, Mancini M. Polyglutamine-expanded androgen receptors from aggregates that sequester heat shock proteins, proteasome components and SRC-1 and are suppressed by the HDJ-2 chaperone. Hum Mol Genet 8, 731-741, 1999 [ Links ]
107. Ellerby L, Hackam A, Propp S, Ellerby H, Rabizadeh S, Cashman N, Trifiro M, Pinsky L, Wellington C, Salvesen G, Hayden M, Bredesen D. Kennedy's disease:caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J Neurochem 72, 185-95, 1999 [ Links ]
108. Yapijakis C, Kapaki E, Boussiou M, Vassilopoulos D, Papageorgiou C. Prenatal diagnosis of X-linked spinal and bulbar muscular atrophy in a Greek family. Prenat Diagn 16:262-65, 1996 [ Links ]
109. Irvine R, Yu M, Ross R, Coetzee G. The CAG and GGC microsatellites of the androgen receptor gene are in linkage disequilibrium in men with prostate cancer Cancer Res 54:28614, 1994 [ Links ]
110. Hardy D, Scher H, Bogenreider T, Sabbatini P, Zhang Z, Nanus D, Catterall J. Androgen receptor CAG repeat lengths in prostate cancer:correlation with age of onset. J Clin Endocrinol Metab 81:4400-05, 1996 [ Links ]
111. Giovannucci E, Stampfer M, Krithivas K, Brown M, Dahl D, Brufsky A, Talcott J, Hennekens C, Kantoff P. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci U S A 94:3320-3, 1997 [ Links ]
112. Jin B, Beilin J, Zajac J, Handelsman D. Androgen receptor gene polymorphism and prostate zonal volumes in Australian and Chinese men. J Androl 21:91-8, 2000 [ Links ]
113. Hardy D, Scher H, Bogenreider T, Sabbatini P, Zhang Z, Nanus D, Catterall J. Androgen receptor CAG repeat lengths in prostate cancer:correlation with age of onset. J Clin Endocrinol Metab 81:4400-05, 1996 [ Links ]
114. Stanford J, Just J, Gibbs M, Wicklund K, Neal C, Blumenstein B, Ostrander E. Polymorphic repeats in the androgen receptor gene:molecular markers of prostate cancer risk. Cancer Res 57:1194-98, 1997 [ Links ]
115. Balic I, Graham S, Troyer D, Higgins B, Pollock B, Johnson-Pais T, Thompson I, Leach R. Androgen receptor length polymorphism associated with prostate cancer risk in Hispanic men. J Urol 168:2245-48, 2002 [ Links ]
116. Ingles S, Ross R, Yu M, Irvine R, La Pera G, Haile RW, Coetzee G. Association of prostate cancer risk with genetic polymorphisms in vitamin D receptor and androgen receptor. J Natl Cancer Inst 89:166-70, 1997 [ Links ]
117. Bennett C, Price D, Kim S, Liu D, Jovanovic B, Nathan D, Johnson M, Montgomery J, Cude K, Brockbank J, Sartor O, Figg W. Racial variation in CAG repeat lengths within the androgen receptor gene among prostate cancer patients of lower socioeconomic status. J Clin Oncol 20:3599-604, 2002 [ Links ]
118. Santos M, Sarkis A, Nishimoto I, Nagai M. Androgen receptor CAG repeat polymorphism in prostate cancer from a Brazilian population. Cancer Detect Prev 27:321-26, 2003 [ Links ]
119. Lange E, Chen H, Brierley K, Livermore H, Wojno K, Langefeld C, Lange K, Cooney K. The polymorphic exon 1 androgen receptor CAG repeat in men with a potential inherited predisposition to prostate cancer. Cancer Epidemiol Biomark Prev 9:439-42, 2000 [ Links ]
120. Cude K, Montgomery J, Price D, Dixon SC, Kincaid RL, Kovacs K, Venzon D, Liewehr D, Johnson M, Reed E, Figg W. The role of an androgen receptor polymorphism in the clinical outcome of patients with metastatic prostate cancer. Urol Int 68:16-23, 2002 [ Links ]
121. Singh Rajender, Lalji Singh, Kumarasamy Thangaraj. Phenotypic heterogeneity of mutations in androgen receptor gene. Asian J Androl 9 (2):147-179, 2007 [ Links ]
122. Huang S, Chou Y, Chang W, Wu M, Yu C, Wu T, Lee Y, Huang J, Wu W, Huang C. Androgen receptor gene polymorphism and prostate cancer in Taiwan. J Formos Med Assoc 102:680-86, 2003 [ Links ]
123. Suzuki H, Akakura K, Komiya A, Ueda T, Imamoto T, Furuya Y, Ichikawa T, Watanabe M, Shiraishi T, Ito H. CAG polymorphic repeat lengths in androgen receptor gene among Japanese prostate cancer patients:potential predictor of prognosis after endocrine therapy. Prostate 51:219-24, 2002 [ Links ]
124. Hsing A, Gao Y, Wu G, Wang X, Deng J, Chen Y, Sesterhenn IA, Mostofi F, Benichou J, Chang C. Polymorphic CAG and GGN repeat lengths in the androgen receptor gene and prostate cancer risk:a population-based case-control study in China. Cancer Res 60:5111-16, 2000 [ Links ]
125. Mishra D, Thangaraj K, Mandhani A, Kumar A, Mittal R. Is reduced CAG repeat length in androgen receptor gene associated with risk of prostate cancer in Indian population. Clin Genet 68:55-60, 2005 [ Links ]
126. Krishnaswamy V, Kumarasamy T, Venkatesan V, Shroff S, Jayanth VR, Paul S. South Indian men with reduced CAG repeat length in the androgen receptor gene have an increased risk of prostate cancer. J Hum Genet 51:254-57, 2006 [ Links ]
127. Correa-Cerro L, Wöhr G, Häussler J, Berthon P, Drelon E, Mangin P, Fournier G, Cussenot O, Kraus P, Just W, Paiss T, Cantú JM, Vogel W. (CAG)nCAA and GGN repeats in the human androgen receptor gene are not associated with prostate cancer in a French-German population. Eur J Hum Genet 7:357-62, 1999 [ Links ]
128. Tayeb M, Clark C, Murray GI, Sharp L, Haites N, McLeod H. Length and somatic mosaicism of CAG and GGN repeats in the androgen receptor gene and the risk of prostate cancer in men with benign prostatic hyperplasia. Ann Saudi Med 24:21-26, 2004 [ Links ]
129. Mononen N, Ikonen T, Autio V, Rokman A, Matikainen M, Tammela T, Kallioniemi OP, Koivisto PA, Schleutker J. Androgen receptor CAG polymorphism and prostate cancer risk. Hum Genet 111:166-71, 2002 [ Links ]
130. Eu Leong Yong, Joyce Lim, Wang Qi, Victor Ong and Amparo Mifsud. Molecular basis of androgen receptor diseases. Ann Med 32:15-22, 2000 [ Links ]
131. Roy A, Lavrovsky Y, Song C, Chen S, Jung M, Velu N, Bi B, Chatterjee B. Regulation of androgen action. Vit Horm 55:309-52, 1999 [ Links ]
132. Denis L, Griffiths K. Endocrine treatment in prostate cancer. Sem Surg Oncol 18:52-74, 2000 [ Links ]
133. Heinlein C, Chang C. Androgen receptor in prostate cancer. Endocr Rev 25:276-308, 2004 [ Links ]
134. van der Kwast T, Schalken J, Ruizeveld de Winter J, van Vroonhoven C, Mulder E, Boersma W, Trapman J.. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int J Cancer 48:189-93, 1991 [ Links ]
135. Newmark J, Hardy D, Tonb D, Carter B, Epstein J, Isaacs W, Brown T, Barrack E. Androgen receptor gene mutations in human prostate cancer. Proc Natl Acad Sci USA 89:6319-23, 1992 [ Links ]
136. Wilding G, Chen M, Gelmann E. Aberrant response in vitro of hormone-responsive prostate cancer cells to antiandrogens. Prostate 14:103-15, 1989 [ Links ]
137. Shi X, Ma A, Xia L, Kung H, de Vere White R. Functional analysis of 44 mutant androgen receptors from human prostate cancer. Cancer Res 62:1496-502, 2002 [ Links ]
138. Miyamoto H, Yeh S, Lardy H, Messing E, Chang C. 5-Androstenediol is a natural hormone with androgenic activityin human prostate cancer cells. Proc Natl Acad Sci USA 95:11083-88, 1998 [ Links ]
139. Yeh S, Miyamoto H, Shima H, Chang C. From estrogen to androgen receptor:a new pathway for sex hormones in prostate. Proc Natl Acad Sci USA 95:5527-32, 1998 [ Links ]
140. Elo J, Kvist L, Leinonen K, Isomaa V, ttu P, Lukkarien O, Vihko P. Mutated human androgen receptor gene detected in a prostatic cancer patient is also activated by estradiol. J Clin Endocrinol Metab 80:3494-500, 1995 [ Links ]
141. Taplin M, Bubley G. Shuster T, Frantz M, Spooner A, Ogata G, Keer H, Balk S. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 332:1393-38, 1995 [ Links ]
142. Yong E, Tut T, Ghadessy F, Prins G, Ratnam S. Partial androgen insensitivity and correlations with the predicted three dimensional structure of the androgen receptor ligand binding domain. Mol Cell Endocrinol 137:41-50, 1998 [ Links ]
143. Gottlieb B, Lehvaslaiho H, Beitel L, Lumbroso R, Pinsky L, Trifiro M. The androgen receptor gene mutations database. Nucleic Acids Res 26:234-38, 1998 [ Links ]
144. Holdcraft R, Braun R. Androgen receptor function is required in Sertoli cells for the terminal differentiation of haploid spermatids. Development 131:459-67, 2004 [ Links ]
145. Singh R, Deepa S, Madhavi S, Gupta N, Chakravarty B, Singh L, Thangaraj K. Male infertility:no evidence of involvement of androgen receptor gene among Indian men. J Androl 27:102-05, 2006 [ Links ]
146. Tut T, Ghadessy F, Trifiro M, Pinsky L, Yong E. Long polyglutamine tracts in the androgen receptor are associated with reduced transactivation, defective sperm production and male infertility J Clin Endocrinol Metab 82:3777-82, 1997 [ Links ]
147. Dowsing A, Yong E, McLachlan R, de Kretser D, Trounson A. Linkage between male infertility and trinucleotide expansion in the androgen receptor gene. Lancet 354:640-43, 1999 [ Links ]
148. Yong E, Loy C, Sim K. Androgen receptor gene and male infertility. Hum Reprod Upd 9:1-7, 2003 [ Links ]
149. Wallerand H, Remy-Martin A, Chabannes E, Bermont L, Adessi G, Bittard H. Relationship between expansion of the CAG repeat in exon 1 of the androgen receptor gene and idiopathic male infertility. Fertil Steril 76:769-74, 2001 [ Links ]
150. Mengual L, Oriola J, Ascaso C, Ballesca JL, Oliva R. An increased CAG repeat length in the androgen receptor gene in azoospermic ICSI candidates. J Androl 24:279-84, 2003 [ Links ]
151. Rajpert-De Meyts E, Leffers H, Petersen J, Petersen J, Andersen A, Carlsen E, Jørgensen N, Skakkebaek N. CAG repeat length in androgenreceptor gene and reproductive variables in fertile and infertile men. Lancet 359:44-46, 2002 [ Links ]
152. Yong E, Liu P, Sim C, Vasan S, Lim P, Ng S. The CAG repeat in the androgen receptor gene is associated with increased risk for either male infertility or prostate cancer (Abstract OR43-3). Proc 80th Ann Meeting Endocr Soc p 108; N Orleans. Bethesda:Endocrine Society; June 24-27, 1998 [ Links ]
153. Wang Q, Ghadessy F, Trounson A, de Kretser D, McLachlan R, Ng S, Yg E. Azoospermia associated with a mutation in the ligand-binding domain of an androgen receptor displaying normal ligand binding, but defective transactivation. J Clin Endocrinol Metab 83:4303-39, 1998 [ Links ]
154. Ghadessy F, Lim J, Abdullah A, Panet-Raymond V, Choo C, Lumbroso R, Tut TG, Gottlieb B, Pinsky L, Trifiro M, Yong E. Oligospermic infertility associated with an androgen receptor mutation that disrupts interdomain and coactivator (TIF2) interactions. J Clin Invest 103:1517-25, 1999. [ Links ]
155. Yong E, Ng S, Roy A, Yun G, Ratnam S. Pregnancy after hormonal correction of severe spermatogenic defect due to mutation in androgen receptor gene. Lancet 344:826-27, 1994 [ Links ]
156. Gottlieb B, Beitel L, Wu J, Trifiro M. The androgen receptor gene mutations database (ARDB):2004 update. Hum Mutat 23:527-33, 2004 [ Links ]
157. Lund A, Juvonen V, Lahdetie J, Aittomaki K, Tapanainen J, Savontaus M. A novel sequence variation in the transactivation regulating domain of the androgen receptor in two infertile Finnish men. Fertil Steril 79:1647-48, 2003 [ Links ]
158. Hiort O, Holterhus P, Horter T, Schulze W, Kremke B, Bals-Pratsch M, Sinnecker G, Kruse K. Significance of mutations in the androgen receptor gene in males with diopathic infertility. J Clin Endocrinol Metab 85:2810-15, 2000 [ Links ]
159. Wang Q, Ghadessy F, Yong E. Analysis of the transactivation domain of the androgen receptor in patients with male infertility. Clin Genet 54:185-92, 1998 [ Links ]
160. Ghadessy F, Lim J, Abdullah A, Panet-Raymond V, Choo C, Lumbroso R, Tut TG, Gottlieb B, Pinsky L, Trifiro M, Yong E. Oligospermic infertility associated with an androgen receptor mutation that disrupts interdomain and coactivator (TIF2) interactions. J Clin Invest 103:1517-25, 1999 [ Links ]
161. Zinn, Ramos P, Elder F, Kowal K, Samango-Sprouse C,Ross J. Androgen receptor CAGn repeat length influences phenotype of 47,XXY Klinefelter syndrome. J Clin Endocrinol Metab 90:5041-46, 2005 [ Links ]
162. Kamischke A, Baumgardt A, Horst J, Nieschlag E. Clinical and diagnostic features of patients with suspected Klinefelter syndrome. J Androl 24:41-48, 2003 [ Links ]
163. Franks S. Polycystic ovary syndrome. N Engl J Med 333:853-61, 1995 [ Links ]
164. Vendola K, Zhou J, Adesanya O, Weil S, Bondy C. Androgens stimulate early stages of follicular growth in the primate ovary. J Clin Invest 101:2622-29, 1998 [ Links ]
165. Chadha S, Pache T, Huikeshoven J, Brinkmann A, van der Kwast T. Androgen receptor expression in human ovarian and uterine tissue of long-term androgen-treated transsexual women. Hum Pathol 25:1198-204, 1994 [ Links ]
166. Schuring A, Welp A, Gromoll J, Zitzmann M, Sonntag B, Nieschlag E, Greb R, Kiesel L. Role of the CAG Repeat Polymorphism of the Androgen Receptor Gene in Polycystic Ovary Syndrome (PCOS). Exp Clin Endocrinol Diab 120:73-79, 2012 [ Links ]
167. Xita N, Georgiou I, Lazaros L, Psofaki V, Kolios G, Tsatsoulis A. The role of SHBG and androgen receptor gene variants in the development of polycystic ovary syndrome. Hum Reprod 23:693-698, 2008 [ Links ]
168. Shah N, Antoine H, Pall M, Taylor K, Azziz R, Goodarzi M. Association of androgen receptor CAG repeat polymorphism and polycystic ovary syndrome. J Clin Endocrinol Metab 93:1939-1945, 2008 [ Links ]
169. Mifsud A, Ramirez S, Yong E. Androgen receptor gene CAG trinucleotide repeats in anovulatory infertility and polycystic ovaries. J Clin Endocrinol Metab 85:3484-3488, 2000 [ Links ]
170. Wang R, Goodarzi M, Xiong T, Wang D, Azziz R, Zhang H. Negative association between androgen receptor gene CAG repeat polymorphism and polycystic ovary syndrome? A systematic review and metaanalysis. Mol Hum Reprod 18:498-509, 2012 [ Links ]
171. Jaaskelainen J, Korhonen S, Voutilainen R, Hippelainen M, Heinonen S. Androgen receptor gene CAG length polymorphism in women with polycystic ovary syndrome. Fertil Steril 83:1724-1728, 2005 [ Links ]
172. Radian S, Baculescu N, Aflorei D, Gussi I, Vladoiu S, Ianas O, Grigorescu F, Coculescu
M. CAG repeat alleles of the androgen receptor are associated with polycystic ovary syndrome (PCOS) in the Romanian population. Endocr Abstr 2010 [ Links ]
173. Mohlig M, Jurgens A, Spranger J, Hoffmann K, Weickert M, Schlösser H, Schill T, Brabant G, Schüring A, Pfeiffer AF, Gromoll J, Schöfl C. The androgen receptor CAG repeat modifies the impact of testosterone on insulin resistance in women with polycystic ovary syndrome. Eur J Endocrinol 155:127-30, 2006 [ Links ]
174. Kim J, Choung S, Choi Y, Yoon S, Kim S, Moon S. Androgen receptor gone CAG repeat polymorphism in women with polycystic ovary syndrome. Fertil Steril 90:2318-2323, 2008 [ Links ]
175. Laisk T, Haller-Kikkatalo K, Laanpere M, et al. Androgen receptor epigenetic variations influence early follicular phase gonadotropin levels. Act Obstetrt Gynecol Scandinav 2010; 89(12):1557-63 [ Links ]
176. Skrgatic L, Baldani D, Cerne JZ, Ferk P, Gersak K. CAG repeat polymorphism in androgen receptor gene is not directly associated with polycystic ovary syndrome but influences serum testosterone levels. J Ster Biochem Mol Biol 128:107-12, 2012 [ Links ]
177. Hickey T, Chandy A, Norman R. The androgen-receptor CAG repeat polymorphism and X-chromosome inactivation in Australian Caucasian women with infertility related to polycystic ovary syndrome. J Clin Endocrinol Metab 87:161-165, 2002 [ Links ]
178. Van Nieuwerburgh F, Stoop D, Cabri P, Dhont M, Deforce D, De Sutter P. Shorter CAG repeats in the androgen receptor gene may enhance hyperandrogenicity in polycystic ovary syndrome. Gynecol Endocrinol 24:669-673, 2008 [ Links ]
179. Dasgupta S, Sirisha P, Neelaveni K, Anuradha K, Reddy A, Thangaraj K, Reddy B. Androgen Receptor CAG Repeat Polymorphism and Epigenetic Influence among the South Indian Women with Polycystic Ovary Syndrome. Plos One 5(8), 2010 [ Links ]
180. Heard E, Clerc P, Avner P. X-chromosome inactivation in mammals. Ann Rev Genet 31:571-610, 1997 [ Links ]
181. Abbout D, Barnett D, Bruns C, Dumesic D. Androgen excess fetal programming of female reproduction:a developmental etiology for polycystic ovary syndrome? Hum Reprod Up 11:357-74, 2005 [ Links ]
182. Li Z, Huang H. Epigenetic abnormality: A possible mechanism underlying the fetal origin of polycystic ovary syndrome. Med Hypotheses 70(3):638-42, 2008 [ Links ]
183. Eisner J, Dumesic D, Kemnitz J, Colman R, Abbott D. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obesity Res 11(2):279-26, 2003 [ Links ]
184. Xu N, Kwon S, Abbott D, Geller D, Dumesic D, Azziz R, Guo X, Goodarzi M. Epigenetic Mechanism Underlying the Development of Polycystic Ovary Syndrome (PCOS)-Like Phenotypes in Prenatally Androgenized Rhesus Monkeys. Plos One 6(11), 2011 [ Links ]
185. Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Pérez-Bravo F, Recabarren S. Maternal serum androgens in pregnant women with polycystic ovarian syndrome:possible implications in prenatal androgenization. Hum Reprod 17:2573-79, 2002 [ Links ]
186. Nestler J. Modulation of aromatase and P450 cholesterol side-chain cleavage enzyme activities of human placental cytotrophoblasts by insulin and insulin-like growth factor I. Endocrinology 121:1845-52, 1987 [ Links ]
187. Barbieri R, Saltzman D, Torday J, Randall R, Frigoletto F and Ryan K. Elevated concentrations of the beta-subunit of human chorionic gonadotropin and testosterone in the amniotic fluid of gestations of diabetic mothers. Am J Obstet Gynecol 154:1039-1043, 1986 [ Links ]
188. Zhu J, Zhu L, Liang X, Xing F, Schatten H, Sun Q. Demethylation of LHR in dehydroepiandrosterone-induced mouse model of polycystic ovary syndrome. Mol Hum Reprod 16:260-66, 2010 [ Links ]
189. Giwercman Y, Xu C, Arver S, Pousette A, Reneland R. No association between the androgen receptor gene CAG repeat and impaired sperm production in Swedish men. Clin Genet 54:435-36, 1998 [ Links ]
190. Edwards A. Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups. Genomics 12:241-53, 1992 [ Links ]
191. Lange E, Sarma A, Ray A, Wang Y, Ho L, Anderson S, Cunningham J, Cooney K. The androgen receptor CAG and GGN repeat polymorphisms and prostate cancer susceptibility in African-American men:results from the Flint Men's Health Study. J Hum Genet 53:220-226, 2008 [ Links ]
192. Vottero A, Stratakis C, Ghizzoni L, Longui C, Karl M, Chrousos G. Androgen receptor-mediated hypersensitivity to androgens in women with nonhyperandrogenic hirsutism: Skewing of X-chromosome inactivation. J Clin Endocrinol Metab 84:1091-95, 1999 [ Links ]
193. Calvo R, Asuncion M, Sancho J, San Millán L, Escobar-Morreale H. The role of the CAG repeat polymorphism in the androgen receptor gene and of skewed Xchromosome inactivation, in the pathogenesis of hirsutism. J Clin Endocrinol Metab 85:1735-40, 2000 [ Links ]
194. Xu N, Azziz R, Goodarzi M. Epigenetics in polycystic ovary syndrome:a pilot study of global DNA methylation. Fertil Steril 94:781-U426, 2010 [ Links ]
195. Heemers H, Tindall D. Androgen receptor (AR) coregulators:A diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev 28:778-808, 2007 [ Links ]

