











 texto em
texto em  Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Los tumores glómicos (TG) o glomangiomas son neoplasias vasculares benignas infrecuentes y surgen de células musculares lisas modificadas del cuerpo glómico que se ubican rodeando las uniones arteriovenosas especializadas en la regulación de la temperatura y el flujo sanguíneo periférico (canales de Sucquet-Hoyer) 1,2.
Estas neoplasias son extremadamente raras y representan alrededor del 2% de todas las neoplasias de tejidos blandos3. Se localizan en zonas acrales periféricas con altas tendencias a afectar las zonas subungueales dérmicas y subdérmicas de los dedos de manos y pies; rara vez, menos del 25%4, se localizan en ubicaciones extracutáneas incluidos estómago, intestino, peritoneo, mediastino, pulmón, tráquea, páncreas, vejiga y vagina2. La mayoría de los TG intraabdominales informados se producen en el estómago; su presentación intestinal es de muy baja incidencia5, y los que involucran específicamente el íleon son sumamente infrecuentes3.
Se presenta el caso de una mujer de 26 años, sin antecedentes personales patológicos ni quirúrgicos de relevancia, que consultó por presentar dolor abdominal crónico de dos meses de evolución, sordo, continuo, de baja intensidad, localizado en región umbilical con irradiación a hipogastrio, sin factores modificadores. No refirió pérdida de peso, fiebre, síntomas uroginecológicos, respiratorios u otra signo sintomatología asociada. El gradual aumento de la intensidad del dolor generó la primera consulta al Servicio de Clínica Médica.
En el momento de la consulta se encontraba en buen estado general, consciente, lúcida, vigil, orientada en tiempo y espacio; hemodinámicamente estable, sin signos de fallo de bomba y con buena perfusión periférica; piel y mucosas normopigmentadas con temperatura y humedad conservadas; buena entrada de aire bilateral sin alteraciones de la mecánica ventilatoria; abdomen plano, blando, depresible, no doloroso a la palpación, con ruidos hidroaéreos conservados, no se palpaban visceromegalias; tacto rectal con esfínter normotónico, paredes de canal anal lisas y elásticas, ampolla rectal libre, dedo de guante limpio. Se realizó laboratorio que informó parámetros hematimétricos, perfiles metabólicos, inmunológicos y proteinograma dentro de valores normales.
Se realizo ecografía, con exploración por vía transabdominal y transvaginal, donde se visualizó ‒ hacia el fondo de saco de Douglas‒ una lesión redondeada de 7×5,7×4 cm, heterogénea, con componente sólido muy vascularizado. En la porción líquida se observaron proyecciones papilares heterogéneas hacia el interior, pero no se logró identificar una conexión con los ovarios a pesar de encontrarse a ambos lados de la lesión. Se continuó la evaluación diagnóstica del tumor pélvico con resonancia magnética nuclear (RMN) con contraste, donde se observaron los ovarios en posición anatómica de tamaño y morfología conservados, y una imagen sólido-quística con componente sólido intensamente vascularizado en fase arterial con irrigación aparentemente dependiente de ramos mesentéricos arteriales en íntimo contacto con asas de intestino delgado (ID). Se realizó tomografía por emisión de positrones (PET-TC) con inyección de 10 mCi de fluorodesoxiglucosa (FDG-F18) y se visualizó en región abdominopélvica una lesión sólido-quística de 6,32×5,92×5,8 cm (Fig. 1), la porción sólida con moderado consumo del marcador y 2,20 SUV (Standardized Uptake Value) máximo; dicha lesión producía el desplazamiento de la vejiga y las asas intestinales adyacentes.
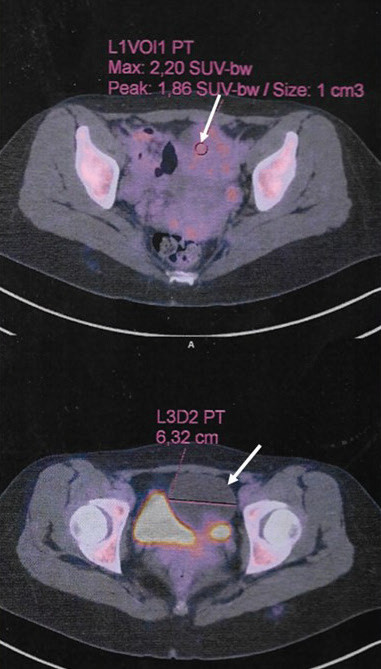
FIGURA 1 Tomografía por emisión de positrones. La flecha muestra el tumor abdominopélvico con captación de 2,20 SUV máximo
Se decidió, entonces, realizar videolaparoscopia diagnóstica con el fin de determinar el origen del tumor. Como consecuencia, se identificó una lesión nodular de aproximadamente 6,5 cm de diámetro en la cara antimesentérica de un asa del intestino delgado (Fig. 2), a 40-50 cm de la válvula ileocecal. Ante la sospecha de tumor de estroma gastrointestinal (GIST) se decidió convertir, y se realizó resección del segmento de intestino delgado comprometido y anastomosis entero-entérica término-terminal por una incisión de Pfannenstiel. La paciente permaneció internada una semana; reinició sus actividades físicas y laborales habituales, al igual que un plan de alimentación balanceado a los 40 días del posoperatorio. Desde entonces lleva 2 años de seguimiento asintomática.
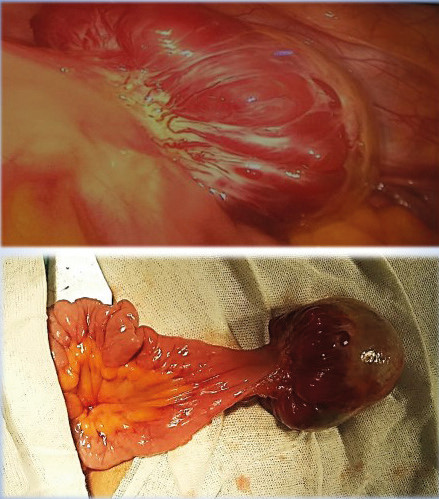
El informe de anatomía patológica arrojó el diagnóstico de tumor glómico de ID, con márgenes libres; los cortes histológicos mostraron la pared del ID con tumoración centrada en la capa muscular propia, con crecimiento predominantemente extramural, bien delimitada y constituida por proliferación de células redondeadas, con núcleos redondeados sin atipias y abundante citoplasma claro. Las células tumorales se disponen rodeando canales vasculares congestivos, algunos hemangiopericitoides. Existen zonas de necrosis, colagenización y hemorragia; no se identificaron mitosis ni pleomorfismo. La serosa y la capa muscular propia adyacentes a la tumoración muestran espacios revestidos por endotelios quísticamente dilatados. La submucosa subyacente al tumor muestra abundantes vasos dilatados y ectásicos.
Por inmunohistoquímica se confirma el diagnóstico: presenta positividad intensa y difusa con vimentina y actina del músculo liso alfa (α-SMA) y negatividad para los restantes marcadores (desmina, CK7, PAX-8, HMB.45, cromogranina y sinaptofisina); con CD34 y CD31 se destaca el endotelio de los canales vasculares, y con c-Kit se identifican frecuentes mastocitos. Los TG suelen ser benignos, pero en ocasiones muestran un tamaño inusualmente grande, crecimiento infiltrativo, necrosis, atipia nuclear y actividad mitótica3,5. La localización extracutánea es poco frecuente; en el tracto gastrointestinal, la mayoría tiene origen gástrico; la localización en el intestino delgado es extremadamente rara.
El TG primario del ID es de muy baja incidencia; al evaluar los informes2,3,4,5,6 se observa que la edad de los pacientes afectados, incluido el nuestro, va desde los 26 hasta los 88 años; no hay informes de casos en edad pediátrica. Afecta a hombres y mujeres en igual proporción (1:1); se han registrado varios casos en 7 países, sin una distribución geográfica predominante.
La presentación del TG en intestino delgado puede ser asintomática, como hallazgo de un estudio de ultrasonografía, o presentar síntomas como dolor abdominal, hemorragia digestiva (hematemesis, melena o hematoquecia), o síntomas asociados a una masa intraabdominal (distensión, vómitos o constipación). En su enfoque diagnóstico se han empleado métodos complementarios como ultrasonografía, endoscopia digestiva, tomografía, resonancia magnética nuclear y tomografía por emisión de positrones; en un caso se procedió directamente a la laparotomía por la sospecha de metástasis de cáncer de recto5.
Por la baja incidencia no se puede afirmar que tenga una localización preferencial en alguna porción en particular del ID (36,4% en duodeno, 18,2% en yeyuno y 45,4% en íleon). La enterectomia parcial de yeyuno o íleon, por laparotomía o laparoscopia, es el tratamiento habitual; en localización duodenal se puede realizar duodenopancreatectomía5; según las comorbilidades de los pacientes se reportaron embolización de la arteria gastroduodenal o mucosectomía endoscópica5.
El diagnóstico previo fue difícil, y solo un caso fue diagnosticado por biopsia endoscópica como TG antes de la cirugía. En otros casos, se diagnosticó como tumor submucoso, carcinoide o neuroendocrino, y otro diagnóstico preoperatorio fue el de GIST (incluso fue nuestro diagnóstico presuntivo).
Los TG del intestino delgado deben diferenciarse de otros citológicamente relacionados como los GIST, los tumores carcinoides, hemangiopericitomas, paragangliomas y linfomas; para ello, los estudios inmunohistoquímicos pueden caracterizar rápidamente el perfil neoplásico; el patrón inmunohistoquímico de los TG casi siempre es positivo para la actina del músculo liso alfa (α-SMA), vimentina y el colágeno tipo IV3,5, y negativa para CD34, c-KIT, citoqueratina, cromogranina y sinaptofisina.
Por lo tanto, los TG deben considerarse como un raro diagnóstico diferencial ante tumores del ID. Por lo general, son benignos y únicos; sin embargo, no hay información de seguimiento a largo plazo, razón por la cual se aconseja su control periódico a efectos de identificar síntomas tempranos de enfermedad.

