











 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
El síndrome de CLOVES (Congenital, Lipomatous, Overgrowth, Vascular Malformations, EpidermalNevi and Spinal/SkeletalAnomalies and/orScoliosisSyndrome) se caracteriza por ser una enfermedad congénita producida por una mutación en el gen PIK3CA.1 Las principales manifestaciones clínicas en estos pacientes son las malformaciones vasculares y de la piel, la presencia de hiperplasia lipomatosa torácica, de tumores y de anomalías viscerales, además de desórdenes neurológicos.2 Este síndrome está incluido dentro de los Síndromes de Sobrecrecimiento Relacionado a PIK3CA (PROS); además de ser poco frecuente, es subdiagnosticado, debido a su complejidad diagnóstica y su similitud con otros síndromes dentro de este grupo, especialmente con el síndrome de Proteus.1,2 Es por ello la importancia de la diferenciación de estas dos patologías, teniendo especialmente en consideración que el síndrome de Proteus está asociado a peor pronóstico.2,3
Presentación del caso
Paciente femenina de 27 años de edad postrada crónica. Presenta al examen clínico los siguientes signos: nevus epidérmico lineal congénito e ipsilateral que se extiende desde el hombro hasta el pie izquierdo (Fig. 1); lipoma en zona poplítea derecha; linfangiomas en antebrazo y en la línea axilar media a la altura del reborde costal (tratados con escleroterapia debido a múltiples sobreinfecciones); hemangiomas en muslo derecho y pie derecho (Figs. 2 y 3); hipertrofia y macrodactilia en ambos pies; y sindactilia en pie izquierdo (Fig. 3). Por lo que, arribamos al diagnóstico de síndrome CLOVES.
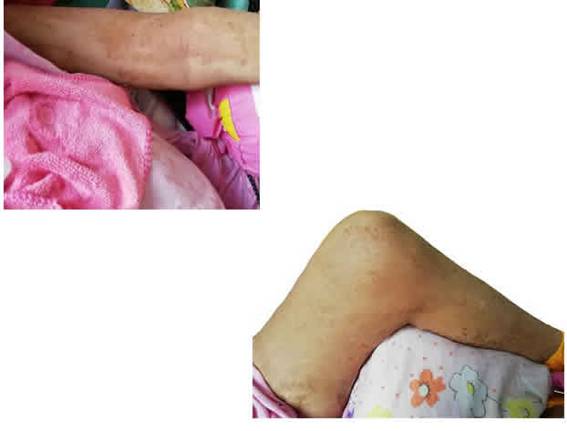


Figura 3: hipertrofia de pies y macrodactilia, pie izquierdo (sindactilia) y pie derecho (hemangioma)
El diagnóstico inicial, durante el periodo neonatal, fue clínico y se propuso el Síndrome de Klippel-Trenaunay, motivado en la existencia de un hemangioma en muslo y pie derecho, e hipertrofia de ambos pies; anomalías que retrasaron el inicio de la deambulación.
Producto de la misma enfermedad, la paciente era evaluada periódicamente en su centro de salud. A los siete años de edad fue examinada por otro grupo de médicos, quienes considerando otros signos congénitos, anteriormente no tomados en cuenta, proponen replantear el diagnóstico. Esta examinación sugirió el diagnóstico de síndrome Proteus, debido a la presencia de un nevus epidérmico lineal y un lipoma en región poplítea derecha.
En la evolución posterior, a los 11 años de edad, la paciente sufre una fractura espontánea de fémur derecho, debido a osteopenia, lo cual ocasiona un acortamiento del mismo. A los 22 años, su dolencia principal se volvió el dolor intenso y persistente localizado en ambos miembros inferiores, que le impide la bipedestación. Asimismo, ha desarrollado cinco episodios de trombosis venosa profunda, tres tromboflebitis y dos tromboembolismos pulmonares; por lo que ha sido hospitalizada frecuentemente.
Durante su última hospitalización, se volvió a replantear el diagnóstico a síndrome de CLOVES, ya que se objetivizó una esplenomegalia sintomática producto de una linfangiomatosis esplénica. Debido a esto, se le realiza una esplenectomía, en la cual, se le extirpa un bazo con dimensiones de 30x25x10 cm y con un peso de 3500 gramos. Además, el órgano presentaba múltiples lesiones quísticas, de entre 0,1cm a 5 cm de diámetro de contenido mucinoso y seroso. Asimismo, en el diagnóstico por imágenes (Fig. 4) se evidenció una escoliosis leve, y en la (Fig. 5) se puede observar la deformidad de los huesos de ambos muslos y piernas.
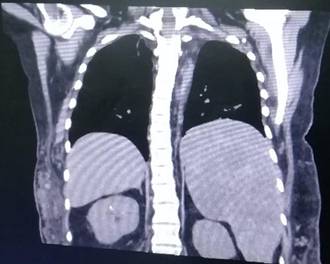
Figura 4: Tomografía de corte coronal donde se observa esplenomegalia con múltiples quistes y escoliosis.

Al momento del presente informe, la paciente se encuentra hospitalizada, con los siguientes objetivos: estudiar su síndrome depresivo, monitorear la terapéutica de anticoagulación oral, y controlar el dolor crónico de los miembros inferiores.
Discusión
Consideramos que el diagnóstico de la paciente fue complejo debido a la baja prevalencia de esta enfermedad; dado la similitud y superposición de sus manifestaciones clínicas con otras patologías clasificadas dentro del espectro de sobrecrecimiento. Dentro de los principales diagnósticos diferenciales están incluidos los síndromes Klippel-Trenaunay-Weber y Proteus.
Uno de los primeros en describirse en la literatura fue el Síndrome de Klippel-Trenaunay-Weber, producido por una mutación del gen PIK3CA, descrito en 1900. Este síndrome se caracteriza por presentar la tríada de “manchas de vino de Oporto”, crecimiento excesivo anormal de tejidos blandos y óseos; y malformaciones venosas.4,5 y 6 Si bien, el síndrome de CLOVES se produce por una mutación en el mismo gen y ambas patologías forman parte del espectro de sobrecrecimientos asociados a PIK3CA ( “PIK3CA- RelatedOvergrowthSpectrum (PROS)” ), este presenta otras manifestaciones clínicas características de tipo progresivas como malformaciones vasculares del tronco, así como diversos grados de escoliosis y estructuras óseas agrandadas; no descritas en el primer síndrome.2
La otra patología con la que se confunde es con el síndrome de Proteus, descrita por primera vez en 1983. A diferencia de los síndromes anteriores, esta se produce por una mutación en el gen AKT1.3,7 Se caracteriza por la presencia de diversos tipos de nevus como el nevus epidérmico lineal y el nevus de tejido conectivo cereberiforme, este último es patognomónico de este síndrome. Además, los pacientes cursan con sobrecrecimiento asimétrico y desproporcionado en extremidades y vísceras (como bazo y timo); y disregulación del tejido adiposo (sobrecrecimiento y lipoatrofia regional)6,9. También; algunos pacientes presentan degeneración pulmonar bullosa, y fenotipos faciales característicos como facie alargada, puente nasal deprimido, dolicocefalia, ptosis menor, entre otras.3,7 y 8 Por otro lado, esta patología se caracteriza por la presencia de tumores a partir de la segunda década de vida, principalmente en ovario y paratiroides; a diferencia de CLOVES. Sin embargo, comparte con CLOVES las malformaciones vasculares de bajo flujo, ya sean capilar, venoso o linfáticas. Asimismo, ambos grupos tienen riesgo de sufrir episodios de tromboembolia pulmonar y terminar en un desenlace fatal.10,11
Otra de las razones por las que fue retador establecer el diagnóstico en el presente caso, se debe a que recién en el 2007 se empiezan a describir casos identificados como síndrome de CLOVES10; y así como en el caso del presente reporte, muchos diagnósticos fueron previamente mal incluidos en otro síndrome de sobrecrecimiento. Como se ha ido mencionando, los pacientes suelen cursar con alteraciones vasculares, dentro de las que destacan las malformaciones linfáticas y venosas de bajo flujo. Además, suelen cursar con compromiso esplénico, ya sea solo esplenomegalia o con la presencia de quistes; como en el caso de esta paciente. En muchos casos, se evidencia compromiso musculoesquelético como en nuestra paciente que presentó escoliosis, asimetría en el crecimiento de los miembros inferiores, macrodactilia y sobrecrecimiento de la planta de los pies. Otro aspecto importante en este síndrome es el compromiso cutáneo, principalmente la presencia de nevus epidérmicos lineales; presentes también en nuestro caso. Así mismo, se observa en esta patología hiperplasia lipomatosa torácica, condición que también tuvo la paciente pero se lo extirparon por sobreinfecciones en múltiples ocasiones. Si bien la literatura menciona que puede cursar con afectación neurológica y en el tracto genitourinario, no se evidenció en la paciente.11
Por último, si bien en la práctica hospitalaria es válido realizar el diagnóstico basado exclusivamente en las manifestaciones clínicas, no es lo ideal en la mayoría de los casos.12 Por ello, sugerimos que cuando haya dificultad para el diagnóstico clínico, se realice un examen genético molecular para conocer la mutación causante del síndrome de sobrecrecimiento. Asimismo, si bien conocer la mutación no mejorará el pronóstico de la enfermedad, permitirá aproximarnos a un valor más real de la prevalencia de casos.
Además, consideramos que es necesario la elaboración de guías de manejo para esta enfermedad crónica. De tal manera que se pueda proporcionar a estos pacientes una atención integral, orientada a asegurar una mejor calidad de vida, manejo del dolor, terapia y rehabilitación física y, un tratamiento de comorbilidades adecuado, enfatizando en el campo de salud mental tanto para los pacientes como para toda la familia.

