











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkResumen y evolución del caso clínico
Niño de 7 años, obeso, con talla baja y retraso del desarrollo psicomotor, que consultó por crisis de tetania por hipocalcemia sintomática, la cual se corroboró con un calcio iónico en 2,5 mg/dL (valor normal [VN]: 4,4-4,9 mg/dL), asociado a la presencia de hiperfosfemia. Dentro de sus antecedentes familiares, se encontró que la madre presentaba obesidad y talla baja, y la hermana de 14 años con talla baja e hipotiroidismo.
Al examen físico se palpó nódulo subcutáneo en cara anterior de muslo derecho por lo que se efectuó ecografía (Figura 1 A). En el pie izquierdo presentaba acortamiento del cuarto y quinto dedo con posición anómala de este último (Figura 1B).
El electrocardiograma mostró un intervalo QT prolongado (Figura 2). Se efectuó ecografía de partes blandas en muslo, que informó calcificación lineal en el tejido celular subcutáneo. La radiografía de extremidades informó hipoplasia tanto de metartasianos como de metacarpianos, con mayor acortamiento del cuarto y quinto metartasiano del pie izquierdo. Se realizó una tomografía computarizada (TC) cerebral que evidenció calcificaciones intracraneales (Figura 3).
En el estudio complementario destacaron los siguientes exámenes: paratohormona (PTH) en 1287 pg/mL (VN: 18-88 pg/mL) 25-hidroxi-vitamina D deficiente, hormonas tiroideas normales, función renal conservada, índice calciuria/creatininuria normal y estudio de síndrome malabsortivo negativo. El electroencefalograma resultó normal y la resonancia nuclear magnética encefálica con hallazgos similares a TC cerebral. La evaluación oftalmológica fue normal.
El paciente recibió terapia con calcio y calcitriol con buena respuesta, logrando el cambio a tratamiento oral en forma satisfactoria. Se configuró así un pseudohipoparatoridismo, se sospechó un fenotipo Ia o Ic de la osteodistrofia hereditaria de Albright (OHA) (MIM: #103580 y MIM: #612462, respectivamente). El estudio del gen GNAS (MIM: #139320) (análisis de la secuencia y prueba de deleción/duplicación, Laboratorio Invitae) evidenció una variante patogénica heterocigota c.139+1G>A (splice donor) lo cual confirmó el diagnóstico.
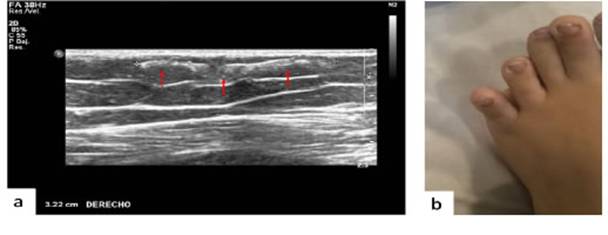
Figura 1: A. Ecografía de partes blandas que muestra imagen hiperecogénica lineal de 3,2 cm de longitud a nivel del tejido celular subcutáneo del muslo derecho. B. Pie izquierdo: acortamiento del cuarto y del quinto dedo con posición anómala del quinto
Se evaluó a la madre en quien se sospechó OHA al evidenciar la presencia de braquidactilia, sumada a sus antecedentes mórbidos. Sin embargo, el estudio del metabolismo calcio-fósforo fue normal. De igual forma, se solicitó estudio a la hermana del paciente. Se citó a toda la familia nuclear para estudio y asesoramiento con genetista, sin embargo, no asisten.
OSTEODISTROFIA HEREDITARIA DE ALBRIGHT
La osteodistrofia hereditaria de Albright (OHA)1 corresponde a un grupo heterogéneo de enfermedades entre las cuales se incluye el pseudohipoparatiroidismo tipo Ia y Ic (subtipos principales y clínicamente idénticos) y el pseudo- pseudohipoparatiroidismo (MIM #612463). Se caracteriza por presentar un fenotipo típico con talla baja, obesidad central, cara redonda, cuello corto, braquidactilia, existencia de osificaciones heterotópicas y retraso mental. Los subtipos Ia y Ic presentan hipocalcemia, hiperfosfemia, incremento de los niveles de PTH, como también resistencia a distintas hormonas como las tiroideas y gonadotrofinas. En el caso del pseudo- pseudohipoparatiroidismo no existe resistencia a la PTH ni a otras hormonas.
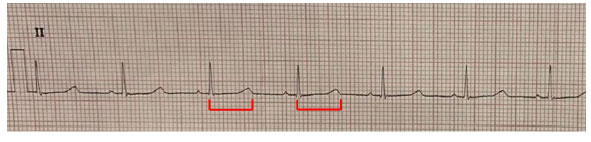
Figura 2: Electrocardiograma que muestra un intervalo QT 0,44 seg, intervalo QT corregido 0,48 seg con frecuencia cardíaca de 72 latidos por minuto
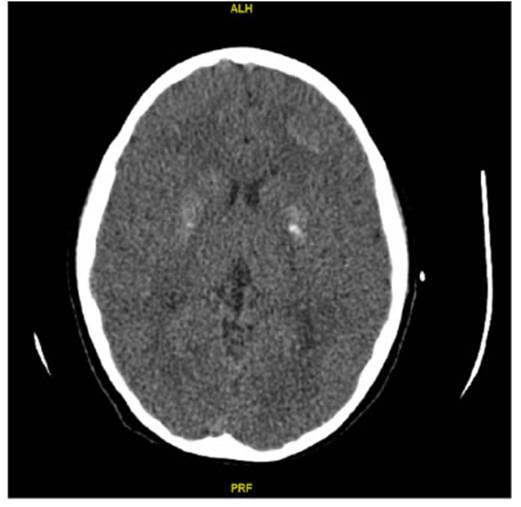
Figura 3: Tomografía computarizada cerebral que muestra calcificaciones parenquimatosas bilaterales y simétricas de sustancia blanca subcortical y cuerpos estriados
La OHA corresponde a una patología autosómica dominante, de variable expresión, debido a una mutación en el locus GNAS (20q13), que codifica para la subunidad alfa de la proteína G (Gs-a), responsable de traducir las señales entre el receptor hormonal y la adenilciclasa, activando la secuencia de señalización intracelular en las células diana. Mutaciones del GNAS generan disminución de la expresión y/o función de la Gs-a lo que se manifiesta clínicamente como OHA con o sin resistencia hormonal.2
Referente a la variante genética que reportamos, c139+1G>A, esta afectaría un sitio donante de empalme en el intrón 1 del gen GNAS, que interrumpiría el empalme del ARN, resultando en un producto proteico alterado o ausente. Esta variante no ha sido descrita previamente en las bases de datos de población, por lo que no se puede estimar la frecuencia de presentación ni asociaciones patogénicas.3 Sin embargo, se han observado alteraciones en este sitio de empalme en individuos con afecciones relacionadas a GNAS, clasificándose esta variante como patogénica.4,5
Respecto a los diagnósticos diferenciales, se puede señalar que la enfermedad de Farber es una lipidosis caracterizada por la tríada clínica de nódulos subcutáneos, artritis con deformación gradual de las articulaciones y compromiso laríngeo con disfonía progresiva. Puede existir retraso del desarrollo psicomotor, pero es infrecuente la presentación con episodios de movimientos corporales de tipo mioclónico o convulsiones. Tampoco se asocia con obesidad o talla baja.
Respecto a la neurocisticercosis, si bien su forma habitual de presentación en zonas endémicas son las crisis convulsivas, es infrecuente la localización del cisticerco en el tejido subcutáneo. Además, los restantes síntomas y signos en nuestro paciente no corresponden a esta infección parasitaria.
Las características principales del complejo de esclerosis tuberosa son la existencia de hamartomas en piel y cerebro con desarrollo de epilepsia refractaria y retraso del desarrollo psicomotor. Sin embargo, los restantes hallazgos en el paciente no eran compatibles.
Finalmente, la poliarteritis nodosa corresponde a una vasculitis de muy variable presentación, en la forma cutánea se destaca la presencia de nódulos dolorosos en extremidades, mialgias, artralgias. Sin embargo, la afectación neurológica es menos frecuente.
El diagnóstico en nuestro paciente fue tardío, situación ocasionada por la escasa adherencia a la atención sanitaria presentada por la familia del niño, como también por el insuficiente seguimiento desde la asistencia primaria de salud.

