











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
La leishmaniosis es una de las principales zoonosis reconocidas a nivel mundial, con potencial fatal para seres humanos y animales y es transmitida mediante picaduras de insectos dípteros hematófagos del género Lutzomyia en el nuevo mundo (Reyes et al. 2015). Los caninos son el principal reservorio del agente causal de la forma visceral en humanos (OPS 2020).
Es causada por parásitos protozoarios del género Leishmania, familia Tripanosomatidae. Los caninos son susceptibles de adquirir la forma visceral (LVc) y la forma cutánea (LCc) (Acero et al. 2015, Cabrera et al. 2021).
El agente causal de esta enfermedad se considera un parásito intracelular obligado de los mamíferos. La infección es endémica en grandes áreas tropicales, subtropicales y en la cuenca del Mediterráneo (del Rosal Rabes et al. 2010, Akhoundi et al. 2017).
Como regla general, esta enfermedad puede corroborarse con rapidez y eficacia por medio de la citología, serología o por la reacción en cadena de la polimerasa (PCR) en perros que muestran signos clínicos evidentes y/o alteraciones graves de parámetros relevantes de laboratorio (Solano-Gallego et al. 2009, Roura 2012).
El diagnóstico parasitológico o examen directo, es considerado el método de referencia (De Los Ríos Alicandú et al. 2010, Srivastava et al. 2011). La sensibilidad del diagnóstico parasitológico o examen directo varía en función de la experiencia del analista, la técnica utilizada para la toma y procesamiento de la muestra, la localización de la lesión, así como del tiempo de evolución de las mismas. Esta técnica requiere de procedimientos invasivos de muestreo lo que implica un mayor riesgo para los animales y es de difícil implementación para su uso generalizado por los programas de salud pública, en los cuales deben ser evaluados un gran número de animales en corto período (OPS 2019).
Las pruebas rápidas de inmunocromatografía disponibles utilizan como antígeno las proteínas recombinantes rK39, que son específicas del complejo L. donovani. Son pruebas cualitativas que detectan anticuerpos en suero, plasma o sangre canina y son las pruebas inmunológicas de rutina más utilizadas por los servicios de la salud de la región (OPS 2019).
Aunque la detección por PCR de LVc utilizando sangre periférica no está exenta de dificultades, presenta ventajas considerables. En particular, su alta sensibilidad en comparación con los métodos convencionales permite la detección de perros asintomáticos, en ocasiones seronegativos y la posibilidad de utilizar muestras menos invasivas (Lachaud et al. 2002).
Actualmente, el diagnóstico de la LVc sigue siendo un problema para los servicios de salud pública, ya que hay una gran variedad de signos y síntomas clínicos que pueden deberse también a otras patologías y la inexistencia de un test diagnóstico que sea 100% especifico y sensible (OPS 2020).
El objetivo del presente estudio fue analizar la utilidad de un diseño de PCR anidada para el diagnóstico de LVc en perros de la ciudad de Corrientes (Argentina).
Materiales y métodos
Muestras. En el Servicio Veterinario de Biología Molecular se recibieron 18 muestras de sangre entera anticoagulada con EDTA de perros de diferentes razas y edades que concurrieron al Hospital Escuela de la Facultad de Ciencias Veterinarias de la Universidad Nacional del Nordeste de la ciudad de Corrientes, con sospecha clínica de padecer leishmaniosis. Las muestras de sangre fueron conservadas en freezer a -20 ºC, por un tiempo no mayor a 30 días hasta su procesamiento. Muestras de la médula ósea de los mismos animales fueron analizadas por el laboratorio de Leishmaniosis de dicha Facultad en busca del parásito utilizando microscopía óptica.
A las muestras se les aplicó el diseño de PCR anidada tomado de Savani et al. (2009), que consta de dos rondas de amplificación sucesivas de una secuencia de ADN que codifica para la subunidad pequeña de ARN ribosomal (Lachaud et al. 2002).
Preparación de ADN. Setecientos microlitros de cada muestra de sangre fueron sometidos a centrifugación durante 4 minutos a 12.000 rpm. El sobrenadante fue descartado, realizando luego la eliminación de glóbulos rojos por lisis osmótica con agua libre de nucleasas (4 veces). Los glóbulos blancos fueron resuspendidos en 500 µl de solución de homogenización conteniendo el detergente CTAB (bromuro de cetil trimetilamonio) e incubado a 60 ºC durante una hora. La purificación de ácidos nucleicos fue realizada por agregado de una mezcla de cloroformo: alcohol isoamílico en proporción 24:1, seguida de precipitación con alcohol isopropílico. La eliminación de sales coprecipitantes fue realizada mediante un lavado con etanol 70% y el precipitado obtenido fue secado a temperatura ambiente y luego resuspendido en 100 µl de agua libre de nucleasas. Las muestras de ADN extraídas fueron conservadas a -20 ºC hasta su utilización.
Amplificación por PCR. Se realizaron dos rondas de amplificación sucesivas (PCR anidada) para identificar al género Leishmania. Las reacciones se llevaron a cabo en 25 µl de volumen final conteniendo: 1X de Buffer de PCR, 2 mM MgCl2; 0,2 μM de cada cebador (S4 y S12 para la primera ronda y S17 y S18 para la segunda, ver Tabla 1); 0,2 mM de una mezcla equimolecular de dNTPs y 2U de Taq ADN polimerasa. La primera ronda se efectuó bajo las siguientes condiciones: desnaturalización inicial: 94 °C 3 min, luego 35 ciclos de desnaturalización a 94 °C 60 s; pegado de cebadores: 50 °C 60 s; extensión: 72 °C 60 s; extensión final: 72 °C 7 min e incubación a 4 ºC hasta su utilización. Los productos obtenidos en la primera amplificación se utilizaron como molde para la segunda ronda que consistió en: desnaturalización inicial: 94 °C 4 min, luego 30 ciclos de desnaturalización a 94 °C 60 s; pegado de cebadores: 55 °C 60 s; extensión: 72 °C 30 s; extensión final a 72 °C 10 min e incubación a 4 ºC hasta su utilización.
Las muestras detectables a Leishmania sp., fueron sometidas a reacciones de amplificación por PCR sencilla para identificación de especies: L. chagasi y L. brasiliensis.
Para la identificación de L. chagasi, se ensayaron dos reacciones de PCR sencillas en 25 μl de volumen final conteniendo 1X de Buffer de PCR, 0,2 μM de cada oligonucleótido cebador (RV1/RV2 o LCS1/LCS3, ver Tabla 1); 0,2 mM de una mezcla equimolecular de dNTPs, 1U de Taq ADN polimerasa y 1,5 o 2,0 mM MgCl2, según se utilicen los pares de cebadores RV1/RV2 dirigidos a una secuencia de ADN altamente repetitiva del ADN kinetoplastídico o LCS1/LCS3 dirigidos a genes de ARNr 18S. Para la amplificación mediada por cebadores RV1/ RV2 se efectuó una desnaturalización inicial a 94 °C 5 min, luego 35 ciclos de desnaturalización a 94 °C 60 s; pegado de primer: 59 °C 60 s; extensión: 72 °C 60 s y una extensión final a 72 °C 5 min e incubación a 4 ºC hasta su utilización. La amplificación mediada por cebadores LCS1/ LCS3 consistió en desnaturalización inicial a 94 °C 2 min, luego 40 ciclos de desnaturalización a 94 °C 30 s; pegado de primer: 55 °C 30 s; extensión: 72 °C 60 s; extensión final: 72 °C 2 min e incubación a 4 ºC hasta su utilización.
Para la identificación de L. braziliensis se ensayaron dos reacciones de PCR sencillas en un volumen final de 25 μl que contuvieron: 1X de Buffer de PCR, 0,2 μM de cada oligonucleótido cebador (b1/b2 o B1/B2, ver Tabla 1); 0,2 mM de una mezcla equimolecular de dNTPs, 1U de Taq ADN polimerasa y 1,5 mM MgCl2. Las condiciones térmicas para la amplificación fueron: desnaturalización inicial: 95 °C 5 min, luego 35 ciclos de desnaturalización a 95 °C 30s; pegado de cebadores: 61 °C 60s; extensión: 72 °C 60s; extensión final: 72 °C 10 min e incubación a 4 ºC hasta su utilización.
En todos los casos el control negativo consistió en 2 µl de agua mientras que las muestras de ADN utilizadas como control positivo fueron cedidas por el Instituto Nacional de Parasitología.
Para la reacción de amplificación del control interno se utilizó un par de oligonucleótidos cebadores dirigidos a un fragmento de ADN mitocondrial específico de la especie canina (ver tabla 1) en un volumen final de 25 µl, conteniendo: 1X de Buffer de PCR, 2,5 mM MgCl2; 0,4 μM de cada Primer; 0,25 mM de una mezcla equimolecular de dNTPs y 1U de Taq ADN polimerasa. En todos los casos se utilizaron 2 µl de ADN e igual volumen de agua como control negativo de PCR. Las condiciones térmicas de amplificación fueron: desnaturalización inicial a 94 °C durante 5 min, seguida de 30 ciclos de desnaturalización a 94 °C durante 45 s; pegado de primer a 58 °C durante 45 s; extensión a 72 °C durante 90 s y extensión final a 72 °C durante 5 min, finalizando con incubación a 4 ºC.
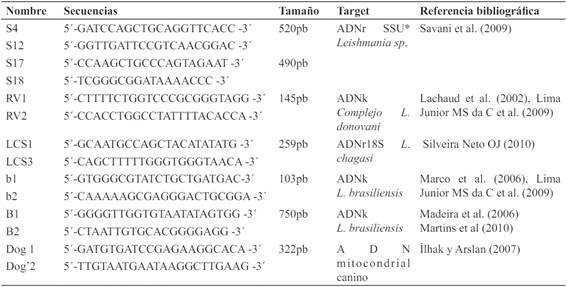
Tabla 1. Secuencias de oligonucleótidos cebadores utilizados en el presente estudio para la amplificación de secuencias específicas.
Electroforesis en gel de agarosa. Los productos de amplificación obtenidos fueron separados por electroforesis en geles de agarosa 2% teñidos con bromuro de etidio y visualizados por transiluminación ultravioleta (UV).
Para la identificación del género Leishmania, los productos de amplificación de ambas rondas son fragmentos de 520 pb y 490 pb, respectivamente.
Para la identificación de L. chagasi los productos de amplificación de ambas reacciones son fragmentos de 145 pb utilizando los cebadores RV1/RV2 y de 259 pb utilizando LCS1/LCS3, para la identificación de L.brasiliensis los productos de amplificación de ambas reacciones son fragmentos de 103 pb utilizando b1/b2 y 750 pb utilizando B1/B2. El control interno corresponde a un amplicón de 322 pb.
Resultados
Las muestras de nueve perros (50%) resultaron detectables por el método molecular observándose bandas de 490 pb, en tanto que las de los otros nueve (50%) fueron no detectables (Figura 1).
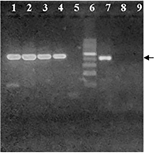
Figura 1 Electroforesis en agarosa 2% de PCRn para identificación de Leishmania sp. (Amplicón específico 490 pb, flecha). Calles 1- 5: muestras, calle 6: marcador de peso molecular (Cienmarker, Biodynamics), calle 7: control positivo, calle 8: control negativo de la primera ronda y calle 9: control negativo de la segunda ronda de amplificación.
En la determinación de especie, L. chagasi fue detectable en el 100% (9/9) de las muestras analizadas (Figura 2), mientras que L. brasiliensis solo fue detectable en el 11% (1/9) de los mismos (Figura 3).
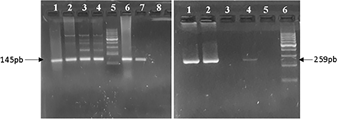
Figura 2 Electroforesis en agarosa 2% de PCR para identificación de Leishmania chagasi. Izquierda: PCR utilizando oligonucleótidos cebadores RV1/RV2 (Amplicón específico 145 pb, flecha). Calles 1-4 y 6: muestras, calle 5: marcador de peso molecular (Cienmarker, Biodynamics), calle 7: control positivo, calle 8: control negativo. Derecha: PCR utilizando oligonucleótidos cebadores LCS1/LCS3 (Amplicón específico 259 pb). Calles 1 y 2: muestras, calle 3: no sembrada, calle 4: control positivo, calle 5: control negativo y calle 6: marcador de peso molecular (Cienmarker, Biodynamics).
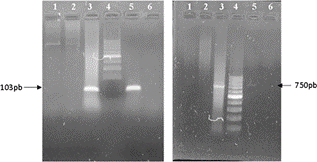
Figura 3 Electroforesis en agarosa 2% de PCR para identificación de Leishmania braziliensis. Izquierda: PCR utilizando los oligonucleótidos cebadores bl/b2 (Amplicón específico 103 pb, flecha). Derecha: PCR utilizando los oligonucleótidos cebadores B1/B2 (Amplicón específico 750 pb). En ambas imágenes, calles 1 - 3 muestras, calle 4: marcador de peso molecular (Cienmarker, Biodynamics), calle 5: control positivo y calle 6: control negativo.
Los resultados obtenidos por PCR anidada sobre las muestras de sangre entera fueron comparados con los obtenidos por el método parasitológico directo informado por el laboratorio de Leishmaniosis de la Facultad de Ciencias Veterinarias de la Universidad Nacional del Nordeste (Tabla 2).
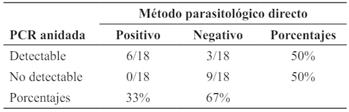
Tabla 2 Resultados obtenidos por método parasitológico directo y PCR anidada en muestras de médula ósea y sangre entera anticoagulada con EDTA de perros.
La sensibilidad del método molecular ensayado fue del 100%, la especificidad del 75%, el valor predictivo positivo del 67% y el valor predictivo negativo del 100%.
En todas las muestras analizadas se visualizó una banda de 322 pb correspondiente al amplicón previsto para el control interno (Figura 4).
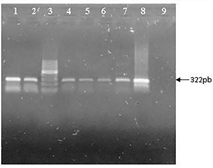
Figura 4 Electroforesis en agarosa 2% de PCR para integridad de material genético utilizando oligonucleótidos cebadores dirigidos a un fragmento de ADN mitocondrial canino (Amplicón específico 322 pb). Calles 1, 2, 4-7: muestras, calle 3: marcador de peso molecular (Cienmarker, Biodynamics), calle 8: control positivo, calle 9: control negativo.
Discusión
En documentos redactados en el Encuentro sobre vigilancia, prevención y control de leishmaniasis visceral en el Cono Sur de Sudamérica en Foz do Iguazú en 2009, se destaca que la LVc es una zoonosis cuya incidencia, letalidad y dispersión geográfica aumentó de manera preocupante en los últimos años en Argentina, Brasil y Paraguay donde se observó un cambio en la epidemiología de la enfermedad que se instaló en áreas urbanas y peri- urbanas con virulencia exacerbada. A nivel regional los perros infectados con o sin manifestaciones clínicas son el principal reservorio de la enfermedad (Dirección de Epidemiología - Área de Vigilancia - SNVS C2 y SIVILA), es por ello que se planteó la necesidad de analizar la importancia y utilidad de pruebas moleculares que pudieran contribuir al diagnóstico de la LVc ya que la vigilancia por laboratorio es parte indispensable de la estrategia de prevención y control integrado de dicha patología.
Usualmente el diagnóstico de LVc está basado en características clínicas y epidemiológicas, como así también en análisis parasitológicos directos y serológicos. La identificación microscópica directa de Leishmania sp. constituye un método simple y económico, aunque posee baja sensibilidad y requiere de un operador entrenado (OPS 2019).
Para fines de vigilancia y estratificación poblacional de LVc los servicios de salud utilizan pruebas inmunológicas, por ser de fácil ejecución, precisión aceptable y por poder aplicarse a gran número de animales simultáneamente en un corto tiempo. Una limitación de esta técnica es que mientras los perros sintomáticos en general producen niveles más elevados de anticuerpos y pueden ser fácilmente detectados, en estadios precoces de infección o animales asintomáticos la sensibilidad de la detección de anticuerpos es menor (OPS 2019). Algunos estudios han demostrado en perros sospechosos de padecer LVc, reactividad cruzada entre Leishmania sp. y Trypanosoma cruzi o Erlichia sp. dependiendo de la prueba serológica. Además, algunos animales pueden tener dificultad para realizar la seroconversión, padecer inmunodeficiencias o ser incapaces de desarrollar una cantidad de anticuerpos que puedan ser detectados en pruebas serológicas. Otra limitación es que no permiten diferenciar infección presente o pasada (da Silva et al. 2004, Caballero et al. 2007, Camargo et al. 2010).
En el presente estudio, se utilizó una técnica de PCR de dos rondas de amplificación sucesivas de una secuencia de ADN que codifica para la subunidad pequeña de ARN ribosomal, lo cual aumenta la sensibilidad, debido a la realización de dos amplificaciones sucesivas y la especificidad al utilizar dos pares de oligonucleótidos cebadores diferentes (Lachaud et al. 2002). Asimismo, durante la etapa de identificación de especies (L. chagasi y L. brasiliensis) se realizaron dos reacciones de amplificación de ADN, utilizando cebadores dirigidos a dos regiones distintas del genoma para cada una, lo cual aumenta la especificidad de la identificación. Los primers RV1/RV2 están dirigidos al fragmento LT1 de mini círculos de ADN kinetoplastídico de L. chagasi, L. infantum y L. donovani; los denominados LCS1/LCS3 están dirigidos a una región específica del gen que codifica para ARNr18S de L. infantum chagasi; b1/b2 y B1/B2 están dirigidos a secuencias diferentes de ADN kinetoplastídico específico de L. brasiliensis. El hecho de utilizar oligonucleótidos cuyos blancos se encuentran en mini círculos de ADN kinetoplastídico del parásito, contribuye a aumentar la sensibilidad de la reacción, dado que se estiman en 10.000 su número por parásito (de Bruijn et al. 1992). Si bien en la muestra detectable para L. brasiliensis se observaron bandas de amplificación específica con ambos pares de oligonucleótidos, la intensidad de las bandas obtenidas con B1/B2 fue menor que con b1/b2 lo cual podría atribuirse a una menor eficiencia en la reacción, aunque este hallazgo también podría relacionarse con el hecho de que amplificaciones de fragmentos mayores, requieren mayor integridad de las moléculas de ADN utilizadas como molde.
Los hallazgos de este estudio fueron contrastados con los resultados del análisis parasitológico obtenido en muestras de médula ósea de los mismos pacientes, realizados por el Laboratorio de Leishmaniosis de la Facultad de Ciencias Veterinarias. La concordancia en los resultados de ambos métodos fue del 83% (15/18 casos) y la discrepancia del 17% (3/18 casos). Seis perros que resultaron positivos por método parasitológico fueron detectables por técnicas moleculares y 9 perros que resultaron negativos por microscopía también lo fueron por PCR anidada. Solo se identificaron tres perros negativos mediante método parasitológico que resultaron detectables por el método molecular. Esta discrepancia podría ser atribuida al menor rango de detección del examen microscópico en cargas parasitarias escasas, a la posible heterogeneidad de la distribución del parásito en la muestra analizada como así también a la mayor sensibilidad de la técnica de PCR anidada para detectar genoma del parásito. Los resultados obtenidos concuerdan con lo publicados por Akhavan et al. (2010), donde la detección de infección por Leishmania mediante PCRn en roedores resultó superior al 50% en las muestras analizadas y del 100% de las muestras que dieron resultado positivo al examen parasitológico directo.
Aunque la reacción de PCR ha demostrado ser un método seguro, en ocasiones puede dar resultados falsos negativos, debido a degradación del material genético durante la conservación, al proceso de extracción de ADN o a la presencia de sustancias inhibidoras. No obstante, para garantizar la calidad de las muestras analizadas en este estudio, se ensayaron reacciones de PCR en las que se amplificó un fragmento de ADN mitocondrial específico de la especie canina, obteniendo bandas de amplificación en todas las muestras analizadas (Landázuri Rafael et al. 2021).
En el presente trabajo no se hallaron muestras en las que el método parasitológico sea positivo y la técnica molecular haya arrojado un resultado no detectable. Andrade et al. (2006) detectaron que el 62,5% de los animales infectados, fueron inicialmente clasificados como no infectados utilizando como pruebas diagnósticas microscopía directa y serología por inmunofluorescencia. Estos autores, al igual que Romero Peñuela et al. (2010) coinciden en que estos métodos usados en áreas endémicas subestiman el número de animales infectados (falsos negativos) permitiendo su permanencia como reservorios. En el presente estudio tres perros fueron negativos en el análisis microscópico pero detectables por PCRn indicando que el último método es útil en la identificación de animales infectados que podrían aun no ser detectados por métodos parasitológicos o serológicos, inclusive en estadios subclínicos.
Es importante tener en cuenta que la reacción de PCR altamente sensible no permite discriminar entre ADN de parásitos vivos o muertos y los productos de reacción pueden obtenerse por amplificación de secuencias de ADN procedente de otros parásitos que pueden estar presentes en perros (da Silva et al. 2004), por lo cual es importante utilizar técnicas que mejoren la sensibilidad y especificidad, tales como la PCR anidada.
De Silva et al. (2022) demostraron un mayor poder de detección de genoma de varias especies de Leishmania utilizando PCR anidada versus PCR sencilla (2,55 fg vs. 25 fg) posibilitando la detección directa del parásito a partir de muestras clínicas sin necesidad de un proceso lento y laborioso como el cultivo del mismo.
En función de nuestros resultados, la reacción en cadena de la polimerasa anidada podría resultar una herramienta útil en el diagnóstico de leishmaniosis canina ya que es independiente de la respuesta inmune del animal, permite analizar la presencia de ADN parasitario en diferentes tipos de muestras y dado que se fundamenta en la amplificación de ácidos nucleicos, en dos rondas sucesivas permite trabajar con pequeños volúmenes de sangre y detectar cantidades mínimas del genoma parasitario (Ferrer y Roura 2010, De Silva et al. 2022), otra ventaja significativa es la utilización de una muestra que requiere procedimientos menos invasivos y riesgosos para su obtención. Debido a que su elevado costo es una limitante, la detección molecular de ADN del parásito podría ser una herramienta de utilidad en casos crónicos, presentaciones tardías y atípicas, como también en aquellos con resultado de examen parasitológico directo negativo o con nexo epidemiológico. Además, el hecho de contar con una técnica molecular de alta sensibilidad y especificidad contribuiría a confirmar el diagnóstico previo a la administración de tratamientos tóxicos y costosos (Deepachandi et al. 2019).
En la LVc, como en otras infecciones, podría ser interesante evaluar la performance (sensibilidad, especificidad, VPP y VPN) de diferentes diseños de PCR; para luego decidir su utilidad en función del objetivo de estudio (técnicas de tamizaje o de diagnóstico) (Lachaud et al. 2002).
La utilización de un método altamente sensible, basado en cebadores dirigidos a ADN repetido en tándem o ADN del kinetoplasto, es una necesidad para la detección óptima y oportuna de todos los portadores de Leishmania sp. y la identificación de especie (Méndez Bejarano 2014).

