











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Se describen tres formas principales de las neurofibromatosis clínica y genéticamente distintas: neurofibromatosis tipos 1 y 2 (NF1 y NF2) y schwannomatosis.
Neurofibromatosis tipo 1 (NF1), conocida como enfermedad de Von Recklinghausen, es el tipo más común. Característicamente presenta múltiples máculas café con leche y neuro fibromas. Se llama NF1 segmentaria cuando las características clínicas se limitan a un área del cuerpo. Es un trastorno genético autosómico dominante con una incidencia de aproximadamente 1 en 2600 a 3000 individuos1,2. Aproximadamente la mitad de los casos son familiares (hereditario). El resto es el resultado de mutaciones de novo (esporádicas)2. Las mutaciones de novo ocurren principalmente en cromosomas derivados del padre, y la probabilidad de NF1 de novo aumenta con la edad paterna avanzada 3. La incidencia de NF1 segmentaria se estima en 1 en 36,000 a 40,000 4. La NF1 se debe a mutaciones en el gen NF1, ubicado en el cromosoma 17q11.2 5. La neurofibromina, el producto proteico codificado por el gen, se expresa en muchos tejidos incluidos el cerebro, el riñón, el bazo y el timo 6. La neurofibromina es un importante regulador negativo de una vía clave de transducción de señales en las células, la vía Ras, que transmite señales mitogénicas al núcleo. La pérdida de neurofibromina conduce a niveles aumentados de Ras activado (unido a GTP), y por lo tanto aumenta la señalización mitogénica, entonces las mutaciones en el gen NF1 dan como resultado la pérdida de producción o la función reducida de la proteína (supresor tumoral), lo que causa un amplio espectro de hallazgos clínicos, incluidos los tumores asociados con NF1 6. La gravedad y las manifestaciones específicas del trastorno varían entre las personas afectadas dentro de la misma familia y de una familia a otra 7.
Neurofibromatosis tipo 2 (NF2) es un síndrome hereditario dominante que predispone a múltiples tumores del sistema nervioso 1-3. Los más comunes son los schwannomas vestibulares bilaterales; los meningiomas intracraneales y espinales y los tumores de la columna vertebral. Las mutaciones en el gen NF2 que se encuentra en el cromosoma 22 4 es la causa de la enfermedad.
El gen NF2 produce merlina, un supresor tumoral también conocido como schwannomina, una proteína relacionada con la membrana celular que actúa como un supresor tumoral 5.6.
Aunque originalmente se pensó que la neurofibromatosis tipo 2 (NF2) era extremadamente rara, el uso de criterios clínicos detallados para identificar estos casos sumado al reconocimiento de que más de la mitad de los casos representan mutaciones de novo y ocurren en ausencia de una historia familiar positiva, así como el uso de pruebas moleculares para identificar mutaciones dentro del gen NF2, han llevado a un mayor reconocimiento de este síndrome1. Algunos estudios, uno de Inglaterra y el otro de Finlandia, encontraron que la incidencia de NF2 puede ser tan alta como 1 en 25,000 8.9.
La schwannomatosis es un trastorno poco común, con una incidencia anual estimada en 0.58 casos por 10000001. Un estudio clínico y patológico a finales de la década de 1990 de pacientes con schwannomas múltiples en ausencia de schwannomas vestibulares, llevó al reconocimiento de que la schwannomatosis era una entidad distinta de NF210. El gen supresor tumoral SMARCB1 sería el predisponente para desarrollar la enfermedad 5 que se caracteriza por presentar múltiples schwannomas no cutáneos en ausencia de schwannomas vestibulares bilaterales.
OBJETIVO
Presentar caso clínico dermatológico de una genodermatosis de escasa consulta en nuestro hospital, lo que constituye una oportunidad diagnóstica familiar ante una enfermedad autosómica dominante con una penetrancia del 100 %, con inferencias de importancia para el resto de la familia afectada.
METODO
Se realizó detección del caso con historia clínica completa, recopilación de estudios complementarios. Búsqueda de bibliografía y correlato de los hallazgos clínicos.
CASO CLINICO
Femenina de 33 años; consulta por lesión en región lumbar izquierda, de varios años de evolución. En los últimos dos meses notó aumento de tamaño, en superficie y en profundidad. Se acompaña de dolor. No refiere antecedentes patológicos propios ni familiares.
Al examen, presenta placa de superficie en empedrado, eritematosa con bordes irregulares, de 11 cm aproximadamente de ancho (Foto N°1). Sobre la placa presenta dos lesiones tumorales de consistencia blanda y compresibles con una pequeña umbilicación central (FotoN°2). La lesión descripta asienta sobre otra lesión macular pigmentada tipo café con leche, de unos 15 cm en su mayor longitud. Se localiza mayormente en región lumbar izquierda. A la palpación, cerca del borde inferior, se palpa un nódulo fusiforme subcutáneo duro, elástico, doloroso, sin adherencias a planos profundos ni superficiales . Al principio nos ocupamos de un síntoma prioritario, el dolor, pues la paciente ha referido limitación de sus actividades por la intensidad de este.

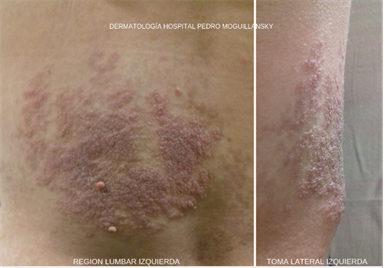
Se solicitó ecografía de partes blandas la cual informó: “Imagen ovoidea de bordes netos, de 18 x 17mm, con eco de estructura sólida, flujo vascular presente, situada en tejido celular subcutáneo compatible con adenopatía.” Fue derivada a cirugía general con indicación de realizar biopsia escisional de piel y de nódulo subcutáneo.
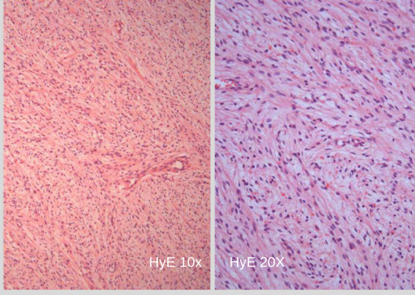
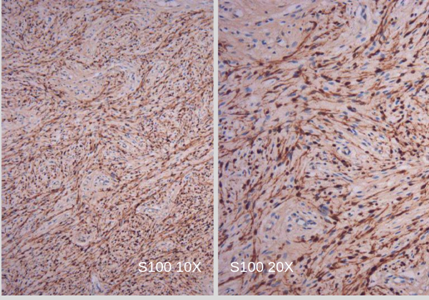
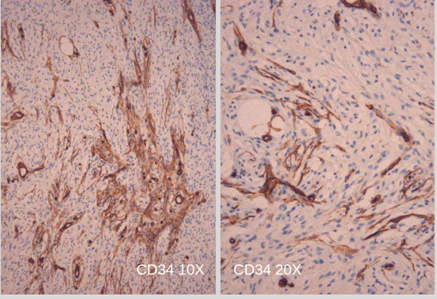
El informe de anatomía patológica de la lesión subcutánea informó: “proliferación de células ahusadas, con mínimo pleomorfismo nuclear y bajo índice mitótico”. (Foto N°3) Planteando diagnóstico diferencial entre fibrosarcoma versus fibromatosis profunda. La inmunomarcación informó: “proliferación fusocelular benigna bien delimitada (Foto N°4); Desmina no reactiva, S100 Positivo (Foto N°5), CD34 Positivo focal (Foto N°6). Hallazgos histológicos e inmunohistoquímicos compatibles con NEUROFIBROMA.

Foto 3 Anatomía patológica. Tinción de hematoxilina y eosina. Células ahusadas (fibroarcoma vs neurofibroma) Dra Picón.
Con el diagnóstico histopatológico se re interroga a la paciente. Refirió tener desde nacimiento una gran mancha café con leche en zona lumbar y en región lateral de cuello y pequeñas manchas en ambas regiones axilares. Surge en interrogatorio que su hermana, papá y tíos paternos también presentan máculas café con leche. Su hija y las hijas de su hermana presentan manchas de iguales características.
Decidimos completar estudios con resonancia magnética de tórax, abdomen y pelvis. Como único dato positivo se mencionan estructuras ganglionares inespecíficas en cadenas ilíacas y femorales bilaterales de aspecto francamente inespecífico.
Fue derivada a oftalmología y neurología. Se citó al resto del grupo familiar
Luego de varios meses concurre a nueva consulta, refiriendo no haber podido completar los estudios y las interconsultas. Describe aumento del dolor lumbar, con algo de impotencia funcional. En esta consulta se toman fotos de la lesión (Foto N°7) y del resto de las lesiones informadas que hacen al diagnóstico clínico de Neurofibromatosis (Fotos N° 8). Evaluada por oftalmología, se objetivó la presencia de más de dos nódulos de Lisch.
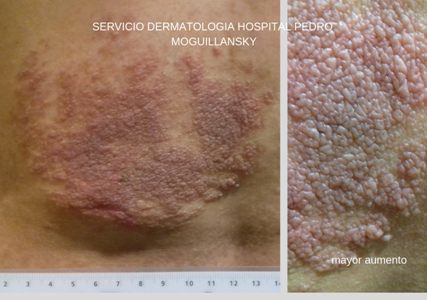
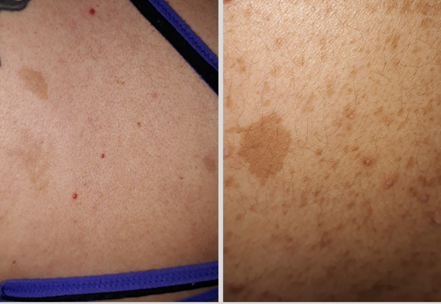
Foto 8. Paciente caso clínico. 33 años. Múltiples máculas café con leche de distinto tamaño y ubicación.
Pudimos evaluar en esta instancia a cuatro miembros de su familia.
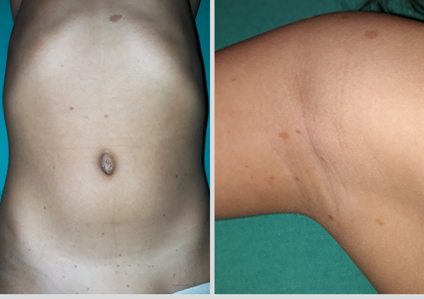
Hija de 5 años: quién presenta múltiples manchas café con leche desde el nacimiento, las cuales fueron aumentando de tamaño (Foto N°9). Sin ningún otro dato relevante en el examen; realizó controles habituales de salud, nunca fue informada respecto a la relación de las manchas con un cuadro patológico.
Hermana de 23 años: se constata la presencia de manchas café con leche, ella describe tenerlas desde el nacimiento. Reconoce el crecimiento de “lunares”, cuyo número aumentó durante los embarazos (Foto N°10). Sabe que toda su familia paterna tiene lesiones similares, pero nunca lo atribuyeron a una situación patológica. Sus tres hijos de 6, 5 y 2 años también presentan múltiples maculas café con leche.
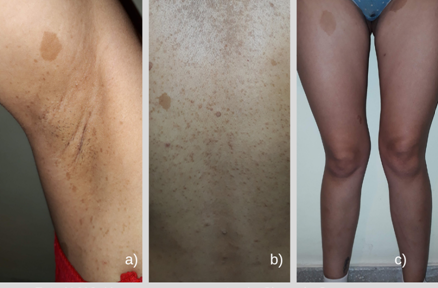
Foto 10. Hermana de la paciente, 23 años. A) Signo de Crowe (pecas intertriginosas). B) Múltiples manchas café con leche y neurofibromas epiteliales. C) Anomalías esqueléticas tibiales.
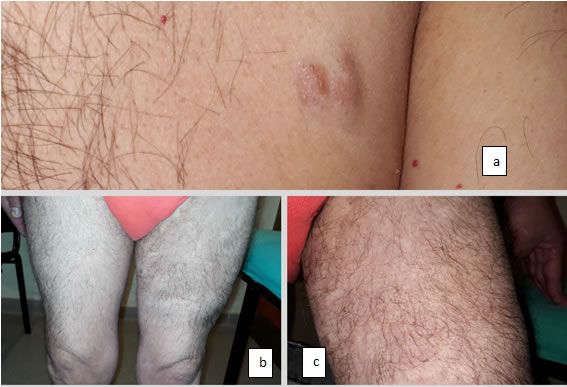
El padre de 65 años presenta múltiples maculas café con leche desde el nacimiento, además de lesiones algo sobre elevadas de consistencia blanda compresibles, compatibles con neuro fibromas epiteliales. Múltiples lesiones nodulares, subcutáneas, de consistencia duro-elástica dolorosas a la palpación en región lateral de abdomen que podrían corresponder a neuro fibromas subcutáneos. En muslo izquierdo presenta una gran lesión tumoral, dando mayor volumen al miembro inferior izquierdo comparado con muslo derecho. Podría corresponder a neuro fibroma plexiforme (Foto N°11). Como antecedente presenta maculopatía pigmentaria desde los 40 años. En evaluación oftalmológica durante esta consulta se objetivó presencia de nódulos de Lisch.
DISCUSIÓN
La Neurofibromatosis 1 rara vez se diagnostica al nacer, porque la mayoría de los signos y síntomas asociados a la enfermedad se desarrollan más tarde. El diagnóstico de NF1 puede realizarse en aproximadamente el 95% de los niños de 6 años afectados, utilizando los criterios diagnósticos del consenso NIH (National Institutes of Health1. Se requieren dos o más criterios para establecer el diagnóstico clínico. Cuadro N°1.

La NF1 es un trastorno multisistémico que afecta principalmente a la piel y sistema nervioso.
Las manifestaciones específicas del trastorno varían entre las personas afectadas, dentro de la misma familia y de una familia a otra.7
Máculas cafés con leche: las máculas se aprecian uniformemente hiperpigmentadas, aparecen durante el primer año después del nacimiento y generalmente aumentan durante la primera infancia. El número de máculas café con leche se estabiliza con el tiempo. No son exclusivas de la NF1 pueden estar presentes en otras genodermatosis.16-18
Pecas axilares o intertriginosas: presentes en niños menores de 6 años, son pequeñas máculas de 1 a 5 mm de diámetro normalmente en zonas no foto expuestas. La localización en axilas se ha denominado signo de Crowe.18 (Foto N°9 y 10).
Neuro fibromas: son tumores benignos de la vaina del nervio periférico que están compuestos por una mezcla de células de Schwann, fibroblastos, células peri neurales y mastocitos. Pueden localizarse en la piel (neuro fibromas cutáneos), a lo largo de los nervios periféricos debajo de la piel o dentro del cuerpo, y a lo largo de las raíces nerviosas adyacentes a la columna vertebral. El número y el tamaño de todos los tipos de neuro fibromas pueden aumentar durante el embarazo. Investigaciones recientes han demostrado que el 75% de los neuro fibromas llevan receptores de progesterona.12
Los neuro fibromas cutáneos son el tipo más común. Consisten en tumores blandos, carnosos, sésiles o pedunculados (Foto N°2). Se mueven con la piel en el examen y no son sensibles.
Algunos se encuentran dentro de la dermis, a la inspección, puede considerarse una placa arciforme, si se logra palpar, y de notarse desarrollo en profundidad, podría denominarse tubérculo; luego del estudio histopatológico confirmando el neurofibroma, su denominación es tumor blando en la piel, a menudo con una coloración violácea suprayacente (Foto N°11 a). El prurito asociado con el crecimiento acelerado de neuro fibromas puede ser un síntoma dominante.5
Neuro fibromas nodulares son lesiones pequeñas que pueden crecer debajo de la piel, como masas firmes y gomosas que pueden ser sensibles o que se presentan más profundamente dentro del cuerpo. Algunos pueden agrandarse hasta el punto en que comprimen las estructuras circundantes o causan dolor, pero no tienden a invadir los tejidos circundantes, como los neuro fibromas plexiformes. Y pueden transformarse en un tumor de estirpe agresiva. 5
Los neuro fibromas plexiformes son una causa importante de morbilidad, y los neuro fibromas plexiformes sintomáticos se asocian con una mayor mortalidad,13 pueden comprimir las vías respiratorias o la médula espinal y pueden transformarse en tumores malignos de la vaina de los nervios periféricos. La característica más común de la transformación maligna de un neuro fibroma plexiforme existente es una lesión dolorosa y en expansión.5
Nódulos de Lisch: son hamartomas del iris, de color tostado. Estos no afectan la visión de ninguna manera. Su hallazgo es útil tanto para establecer un diagnóstico de NF1 en un niño como para determinar si un padre se ve afectado. Estas lesiones se detectan en menos del 10% de los niños afectados menores de seis años, pero se observan en más del 90% de los adultos.
Gliomas de la vía óptica: se producen en el 15% de los niños menores de seis años con NF1.5 Raramente ocurren en niños mayores y adultos. Los gliomas ópticos son típicamente astrocitomas de bajo grado.14 Pueden surgir en cualquier lugar a lo largo de la vía visual anterior a las radiaciones ópticas e involucrar a los nervios ópticos, el quiasma y los tractos ópticos posquiasmáticos. Los síntomas y signos de un glioma de la vía óptica pueden incluir disminución de la agudeza visual o visión del color, función pupilar anormal, proptosis y atrofia del nervio óptico. Aunque los gliomas asociadas a NF1 no progresan a malignidad, los adultos jóvenes con NF1 son propensos al desarrollo de gliomas malignos. 17
Displasia ósea larga y pseudoartrosis: las anomalías óseas en NF1 incluyen pseudoartrosis y displasia ósea, que forman parte del Consenso de los Institutos Nacionales de Salud de los Estados Unidos (NIH) la displasia ósea de huesos largos se presenta típicamente en lactantes o niños pequeños como arqueamiento anterolateral de la tibia, que progresa a estrechamiento del conducto medular, engrosamiento cortical y fractura. Pueden presentar también baja estatura, escoliosis, fibromas no osificante y osteoporosis.12 14
Describimos aquí solo las lesiones incluidas en los criterios diagnósticos, pero cabe destacar que son múltiples los signos y síntomas acompañantes multisistémicos
Según un estudio aproximadamente el 46% de los casos esporádicos de NF1 no cumplen con los Criterios de diagnóstico de NIH antes de 1 año. Casi todos los pacientes con NF1 cumplen con los criterios de diagnóstico a los 8 años, y todos lo hacen a los 20 años. El orden usual de aparición de las características clínicas enumeradas como criterios NIH es máculas cafés con leche, pecas axilares, nódulos de Lisch y neuro fibromas. El glioma óptico sintomático generalmente se diagnostica a los 3 años, y las lesiones óseas características suelen aparecer durante el primer año de vida. 15
CONCLUSION
La neurofibromatosis es una genodermatosis de amplia variedad clínica, pero de características distintivas. En nuestra paciente pudimos constatar 6 de los 7 criterios diagnósticos, pero de forma retrospectiva, ya que en la consulta inicial dominó el síntoma dolor y no fue advertida la multiplicidad de signos clínicos presentes. Lo importante de este caso para nosotros es que fue una importante oportunidad para la familia obtener el diagnóstico, lograr los controles y seguimientos interdisciplinarios, sobre todo para los miembros menores de la familia a quienes podrán detectar precozmente las distintas complicaciones de la enfermedad y obtener un asesoramiento genético, por tratarse de una enfermedad autosómica dominante con alta penetrancia. En cuanto a la referencia bibliográfica pudimos correlacionar la mayoría de las características de la enfermedad al poder evaluar a cuatro miembros afectados de una familia con distintas edades y distintas manifestaciones clínicas de la misma enfermedad.

